新研究揭示PD-1通路调节ILC2代谢,PD-1激动剂治疗可改善气道高反应性
最近在Nature communication上线了一篇标题为 “PD-1 pathway regulates ILC2 metabolism and PD-1 agonist treatment ameliorates airway hyperreactivity” 的文章。该文的通讯作者是来自美国南加州大学凯克医学院分子微生物学与免疫学学系的Omid Akbari。
过敏性哮喘是一种与呼吸系统症状有关的慢性疾病,如呼吸短促、胸闷和咳嗽火性气道疾病。
近几十年来,哮喘的流行率显著增加,全世界约有3.39亿人受到影响。因此,确定新的治疗靶点已成为确保该病有效控制的必要。哮喘的动物模型可以很好地用来模拟哮喘的临床特征,主要是气道高反应性(AHR),它依赖于T-helper 2 (Th2)细胞因子的分泌;白介素(IL)-5促进嗜酸性粒细胞进入肺,而IL-13诱导杯状细胞增生和粘液生成。虽然Th2细胞被公认为2型炎症的关键控制者,但2型先天淋巴样细胞(ILC2s)的早期激活已成为哮喘启动和放大的关键步骤.
PD-1是B7/CD28家族成员,主要表达在活化的T细胞、B细胞和巨噬细胞上。配体结合PD-1, PD-L1 PD-L2,紧随其后的是一连串的胞内信号,导致T细胞抑制和疲惫,PD-1胞质域进行磷酸化的酪氨酸残基,促进招聘的蛋白质酪氨酸磷酸酶(中),主要是Src同源区域2 domain-containing phosphatase-2 (SHP-2)。这随后导致细胞存活和增殖所必需的几种激酶的去磷酸化。由于PD-1通路具有恢复T细胞功能的能力,阻断PD-1通路已成为多种肿瘤的治疗策略。目前美国食品和药物管理局(FDA)批准了6种PD-1途径抑制剂,用于16种肿瘤类型。相反,PD-1缺乏与小鼠的自我耐受性改变和自身免疫性疾病有关。因此,PD-1轴作为治疗各种炎症和自身免疫性疾病的潜在治疗靶点受到了广泛关注
技术路线:

一、PD-1在IL -33激活的ILC2中被高度诱导
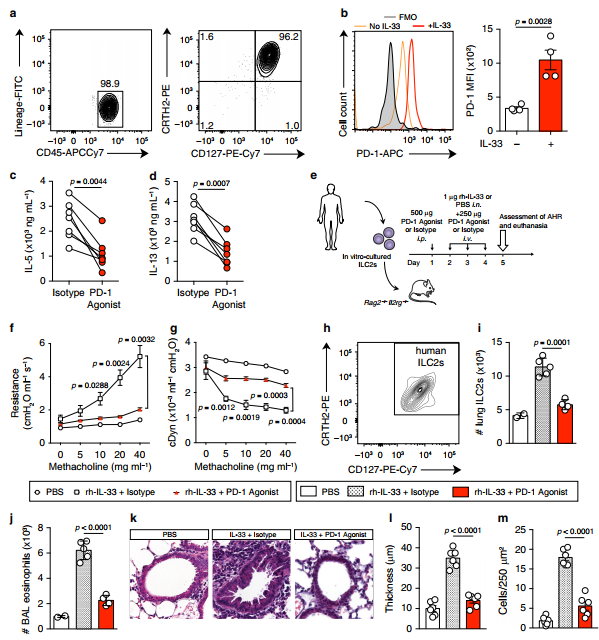
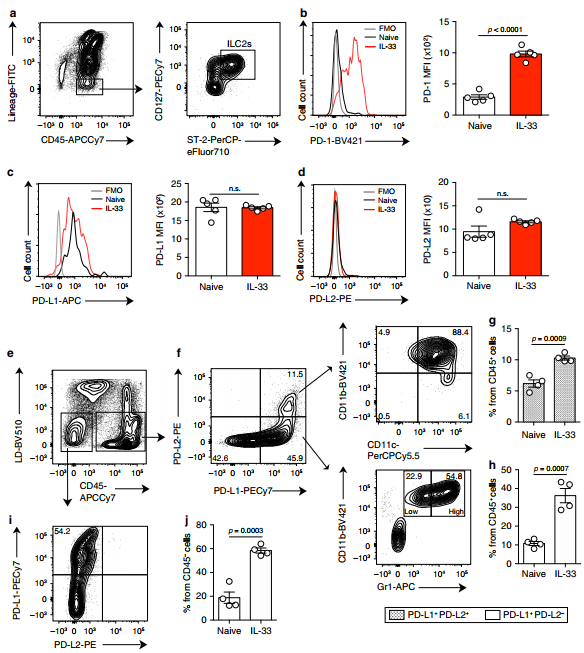
我们首先对PD-1在肺ILC2s上的表达进行了表征,流式细胞术鉴定为CD45+谱系- ST2+ CD127+。在整个研究过程中,未转染小鼠的ILC2s被定义为naive (nILC2s), il -33转染小鼠的ILC2s被定义为activated (aILC2s)。PD-1仅在很少的nILC2s群体中表达;然而,经鼻刺激IL-33可显著诱导PD-1的表达。我们还评估了PD-L1和PD-L2在肺ilc2上的表达。PD-L1在naive和aILC2s中均高表达,而PD-L2不表达。抑制信号的产生和传递需要PD-1与其配体之间的相互作用。为了确定在我们的ilc2依赖性哮喘模型中PD-1配体的潜在来源,我们对PD-L1和PD-L2在CD45+和CD45活肺细胞上的表达进行了表征。
在CD45+细胞中发现了两个阳性群体:(i)同时表达PD-L1和PD-L2的群体,主要代表CD11b+ CD11c+细胞;(ii)仅表达PD-L1的群体,主要代表CD11b+ Gr-1+细胞。有趣的是,在IL-33诱导下,这两个人群的百分比显著增加。与此同时,CD45细胞群体也能表达和上调PD-L1的表达,以应对IL-33。
综上所述,这些结果表明PD-1对ILC2s具有高度诱导作用,免疫和非免疫肺群体可在ILC2依赖性哮喘中提供PD-1配体来源。
二、PD-1在aILC2s中调节细胞因子的产生和存活
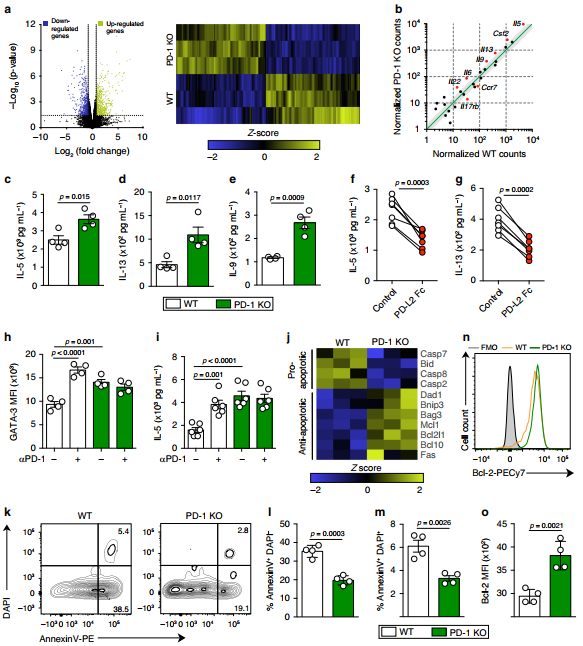
PD-1在肺aILC2s中显著表达,我们使用RNAsequencing分析比较了从野生型(WT)和PD-1敲除(KO)小鼠中经facs分类的aILC2s的转录谱。有趣的是,PD-1缺失导致aILC2s中有840个显著调控基因:426个基因上调,414个基因下调。
比较了细胞因子编码基因在中的表达。l5、Il13、Il9和Csf2在没有PD-1的情况下高度上调(图2b)。
WT和PD-1 KO小鼠aILC2s培养24 h,定量培养上清细胞因子。ILC2的激活导致Th2细胞因子的分泌; 然而,PD-1 KO aILC2s显示出更多的IL-5、IL-13和IL-9(图2c e)。
确定PD-L2是否会抑制Th2细胞因子的产生。PD-L2 Fc使IL-5和IL-13的分泌减少了约2- 3倍(图2f, g)。
PD-L1的组成性表达是否代表了ILC2s中功能PD-1配体的来源。在没有PD-1配体来源,以及PD-1阻断抗体或同型对照的情况下,用IL-33在体外刺激nILC2s。
PD-1阻断后,关键转录因子GATA-3的表达和IL-5的产生显著增加(图2h, i)。这些结果表明,在ILC2s中抑制了潜在的PD-1/PD-L1相互作用。
与WT aILC2s相比,PD-1 KO中Bid、Casp2等促凋亡基因表达明显下调,而Bcl2l1、Bag3、Mcl1等抗凋亡基因表达上调。
AnnexinV+ DAPI和双阳性细胞在PD-1 KO aILC2s中均显著降低,表明细胞凋亡和死亡减少(图2k m)。
流式细胞仪检测凋亡抑制蛋白Bcl-2的表达。PD-1 KO aILC2s中Bcl-2的表达显著且高于WT aILC2s(图2n, o)。
综上所述,这些结果表明PD-1轴积极参与ILC2存活的调控。
三、PD-1的缺乏增强了aILC2s的糖酵解代谢
有氧糖酵解被认为是T细胞激活和增殖的标志。虽然氨基酸和需氧糖酵解可能对ILC2s31至关重要,但ILC2代谢似乎更依赖于脂肪酸(FA)。
PD-1缺陷导致糖酵解基因(如Hk1、Pkm和G6pdx)显著上调,以及FA代谢相关基因的下调。PD-1 KO aILC2s中谷氨酰胺依赖基因也被上调(图3a)。
用荧光葡萄糖模拟物2- NBDG和流式细胞术来测量活细胞中葡萄糖掺入的程度。根据转录分析,在aILC2s中,PD-1缺失导致葡萄糖摄取增加,提示葡萄糖代谢差异和葡萄糖消耗增加(图3b)。
PD-1 KO ILC2s与WT ILC2s相比,在IL-33的作用下,主要的葡萄糖转运体glut1的表达上调(Supplementary Fig. 2B)。
代谢组学进一步用于评估PD-1缺乏对代谢产物的影响。PD-1 KO aILC2s与WT aILC2s相比,糖酵解和磷酸戊糖途径的代谢物水平显著升高。这些代谢物包括磷酸甘油酸、磷酸正丁糖、磷酸戊糖和磷酸己糖(图3c f)。
为了证实不同的2-NBDG摄取和代谢组学结果反映了糖酵解代谢的功能差异,我们测量了耗氧率(OCR)、氧化磷酸化反应(OXPHOS)和细胞外酸化率(ECAR)。PD-1 KO aILC2s表现出备用呼吸能力的下降,说明PD-1的缺乏触发了从OXPHOS到有氧糖酵解的转换(图3g)。
在这种情况下,葡萄糖发酵成乳酸,而不是氧化线粒体由于高能量需求。此外,PD-1 KO的细胞能量表型分析显示ECAR大于WT aILC2s,这意味着在最大呼吸时OCR/ECAR比值较低,糖酵解能力较高(图3h, i)。
这些结果共同揭示了PD-1缺乏与aILC2s中葡萄糖需求增加相关,促使代谢显著向糖酵解方向转变

四、PD-1抑制aILC2s中蛋氨酸和谷氨酰胺分解代谢
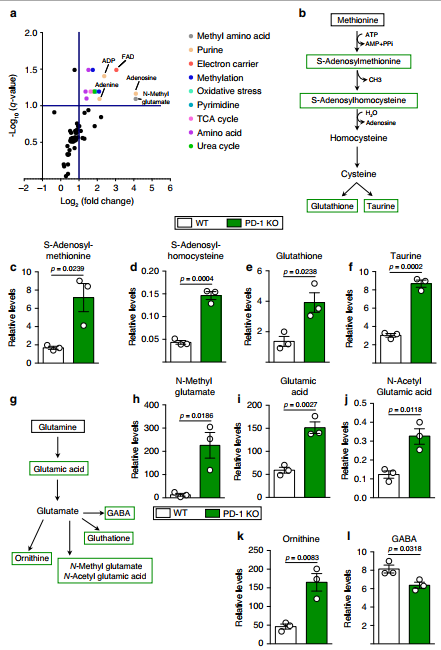
氨基酸分解代谢产生蛋白质合成的代谢物,但也可以作为信号分子控制免疫细胞生长、核苷酸合成、氧化还原控制和许多其他功能35。我们的代谢组学分析显示PD-1的缺乏通常会改变aILC2s的代谢活性。特别是,与嘧啶和嘌呤合成有关的中间代谢物,如5 -二磷酸腺苷(ADP)和腺嘌呤,以及甲基化有关的中间代谢物的上调(图4a)。
第一个途径是蛋氨酸途径(图4b)。两个重要中间代谢物的相对量蛋氨酸代谢,S-Adenosylmethionine (SAM)和SAdenosylhomocysteine (SAH),在PD-1 KO aILC2s显著增加(图。4 c, d)。
两端蛋氨酸代谢产物的分解代谢,谷胱甘肽和牛磺酸,表现出更高的相对量PD-1 KO aILC2s WT aILC2s相比(图4 e, f)。
在筛选的途径中,谷氨酰胺分解也受到影响(图4g)。谷氨酰胺的消耗是关键的免疫细胞代谢,特别是淋巴细胞增殖和细胞因子的生产。
PD-1的缺乏增加了谷氨酰胺代谢中几个中间产物和末端代谢物的相对水平,如谷氨酸、n -甲基谷氨酸、n -乙酰谷氨酸(NAG)和鸟氨酸(图4h k)。
与WT aILC2s相比,只有GABA的相对含量在PD-1 KO中显著降低(图4l)。
综上所述,这些结果表明PD-1缺乏增强了aILC2s的氨基酸分解代谢,特别是依赖蛋氨酸和谷氨酰胺的代谢途径。
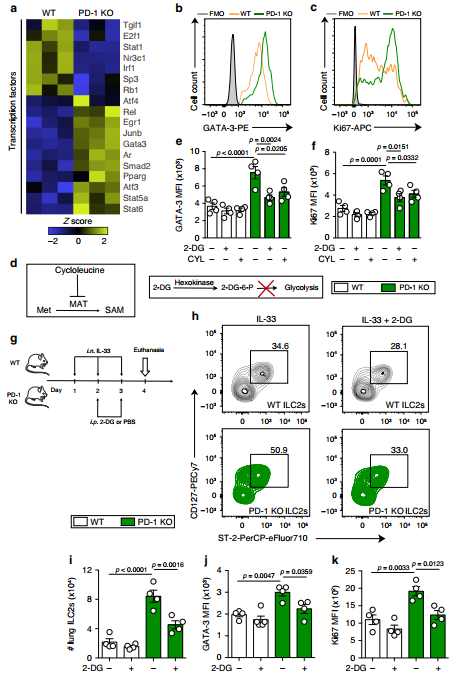
五、PD-1通过代谢调控抑制aILC2的增殖。
PD-1缺陷上调了Th2分化信号通路的相关基因,包括Gata-3、Junb、Stat5a和Stat6,下调了参与Th1分化和激活的基因,如Irf1和Stat1(图5a)。
根据这些结果,通过流式细胞仪检测,缺乏PD-1的aILC2s中,GATA-3的表达强度更高(图5b)。
Ki67核内染色显示,在没有PD-1的情况下,活的aILC2s具有高度增殖能力(图5c)。为了评估PD-1 KO ILC2s的代谢转移与增殖之间的因果关系,我们分别使用竞争性抑制剂2-deoxy-d-glucose (2-DG)和cycloleucine (CYL)抑制糖酵解和蛋氨酸分解代谢。2-DG形成不能进行进一步糖酵解的2-DG-6- p,而CYL抑制催化蛋氨酸转化为SAM的蛋氨酸腺苷转移酶(MAT)(图5d)。
相对较低浓度的这些抑制剂足以显著降低PD-1 KO aILC2s中GATA-3的表达和增殖,而对WT aILC2s的影响较弱或不显著(图5e, f)。
为了将我们之前的观察结果与背景相结合,我们研究了体内糖酵解抑制对aILC2s的影响。如图5g所示,在经鼻注射IL-33的同时,腹腔注射抑制剂2-DG。
PD-1 KO小鼠肺部ILC2s数量高于WT小鼠。有趣的是,在PD-1 KO小鼠中,2-DG显著降低了ILC2百分比(CD45+,细胞系细胞)和绝对计数,但在WT小鼠中没有(图5h, i)。
符合这些结果,2 dg治疗导致明显降低GATA-3 PD-1 KO肺aILC2s Ki67的表情,虽然很弱影响WT aILC2s(图5 j, k)。
综上所述,体外和体内实验表明,PD-1不足与对糖酵解代谢转变,从而提高ILC2活化和增殖潜力IL-33-induced哮喘
六、ILC2s上PD-1的表达可改善AHR和肺部炎症
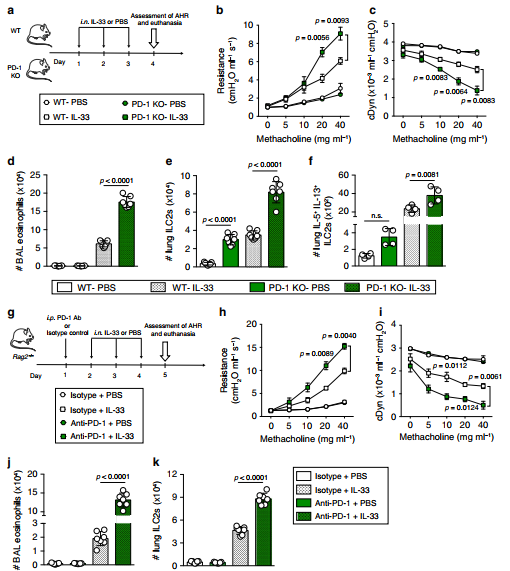
在最后一次鼻内刺激一天后,使用FinePointe RC系统(Buxco Research Systems)对麻醉的气管造口小鼠的肺阻力和动态顺应性(cDyn)进行直接测量,然后进行支气管肺泡灌洗(BAL)和肺组织样本分析(图6a)。
经鼻给予IL-33可显著增加WT和PD-1 KO小鼠的肺抵抗;然而,il -33处理的PD-1 KO小鼠的肺抵抗力明显高于il -33处理的WT小鼠(图6b)。与肺阻力一致的是,il -33处理的PD-1 KO的动态顺应性结果显示,与il -33处理的WT小鼠相比,其响应更低(图6c)。
PD-1是il -33诱导的AHR的负调控因子。虽然IL-33处理显著诱导WT和PD-1 KO小鼠BAL嗜酸性粒细胞增多,但IL-33处理的PD-1 KO小鼠的嗜酸性粒细胞数量明显高于WT小鼠(图)。6 d)。
同样,在IL-33刺激后,PD-1 KO小鼠的肺中ILC2s的数量更高,甚至在幼稚小鼠中,IL-5+ IL-13+ ILC2s的数量也更高(图6e, f)。
第5天测定AHR、BAL嗜酸性粒细胞和肺ILC2值(图6g)。
与对照组相比,抗pd -1处理的Rag2 /小鼠在增加乙酰胆碱浓度、降低动态顺应性、更高的嗜酸性粒细胞招募和更高的肺ILC2数量方面表现出更大的肺抵抗(图6h k)。
七、PD-1激动剂降低人源化小鼠的ilc2依赖性AHR。
由于PD-1在自身免疫性疾病和我们的哮喘模型中发挥有益的调节作用,我们开发了一种人类PD-1激动剂,用于靶向AHR中的PD-1+ILC2s。人类ILC2s被鉴定为CD45+谱系CD127+ CRTH2+(图7a)。
首先,我们观察到PD-1在重组人(rh)- IL-2和rh- il -7培养的人ILC2s上表达,并对rh- il33产生重要的诱导反应(图7b)。
体外检测了PD-1激动剂对人ILC2s产生Th2细胞因子的影响。有趣的是,与同型相比,PD-1激动剂强烈抑制了IL-5和IL-13的产生(图7c, d)。
在最后三天内,用PBS或rh-IL-33对小鼠进行鼻内刺激以诱导AHR(图7e)。
值得注意的是,PD-1激动剂处理的小鼠与同型处理的小鼠相比,肺抵抗显著降低(图7f)。
与肺抵抗一致,PD-1激动剂治疗小鼠的动态顺应性表现出更高的反应(图7g)
激动剂处理的小鼠肺部炎症消失,人类ILC2s数量减少,嗜酸性粒细胞减少,气道上皮厚度减少(图7h m)。
总的来说,这些研究首次证实PD-1的激动性激活抑制了IL-33和hdm诱导的哮喘模型中的AHR,并为哮喘性过敏提供了一种新的特异性治疗方法。