非编码RNA编码蛋白在肿瘤中的作用
lncRNA一般被认为是没有编码蛋白能力的RNA,但是近期研究证明非编码RNA具有编码蛋白的能力,并在肿瘤中发挥重要作用。今天来讲一篇关于lncRNA通过编码蛋白质促进结肠癌(CRC)进展的文章,题名为:Small Protein Hidden in lncRNA LOC90024 Promotes "Cancerous" RNA Splicing and Tumorigenesis,发表在Advanced Science期刊上,IF=15.84。
技术路线:
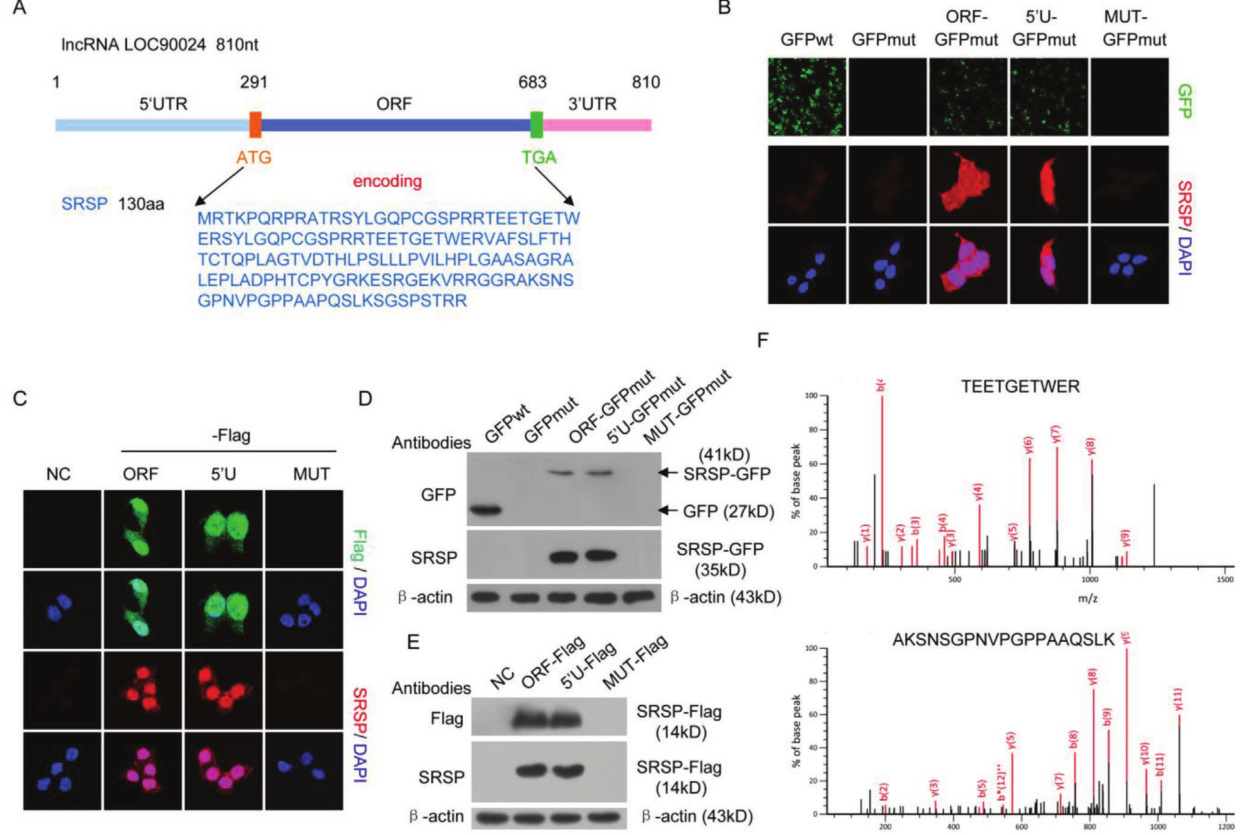
作者首先使用了核糖体RIP对结合核糖体的RNA进行纯化,并通过RNA测序技术分析显示lncRNA LOC90024可以与核糖体结合,这表明LOC90024可能以lncRNA分子的形式翻译为蛋白质/肽或参与翻译调控。生物信息学分析表明,LOC90024中存在一个393个核苷酸的ORF,可能编码130个氨基酸的小蛋白。将这种小蛋白命名为SRSP。同源性分析表明,SRSP在灵长类动物(中高度保守,但在SRSP中没有经典的结构域,这表明SRSP是一种功能未知的新型蛋白质。为了确认lncRNA LOC90024中的预测ORF可以翻译,将预测ORF中的起始密码子ATG突变为ATT。突变后消除了SRSP-GFP或SRSP-Flag融合蛋白的表达。通过质谱进一步鉴定了SRSP-GFP蛋白。总的来说,这些数据表明lncRNA LOC90024实际上编码一种小蛋白SRSP。
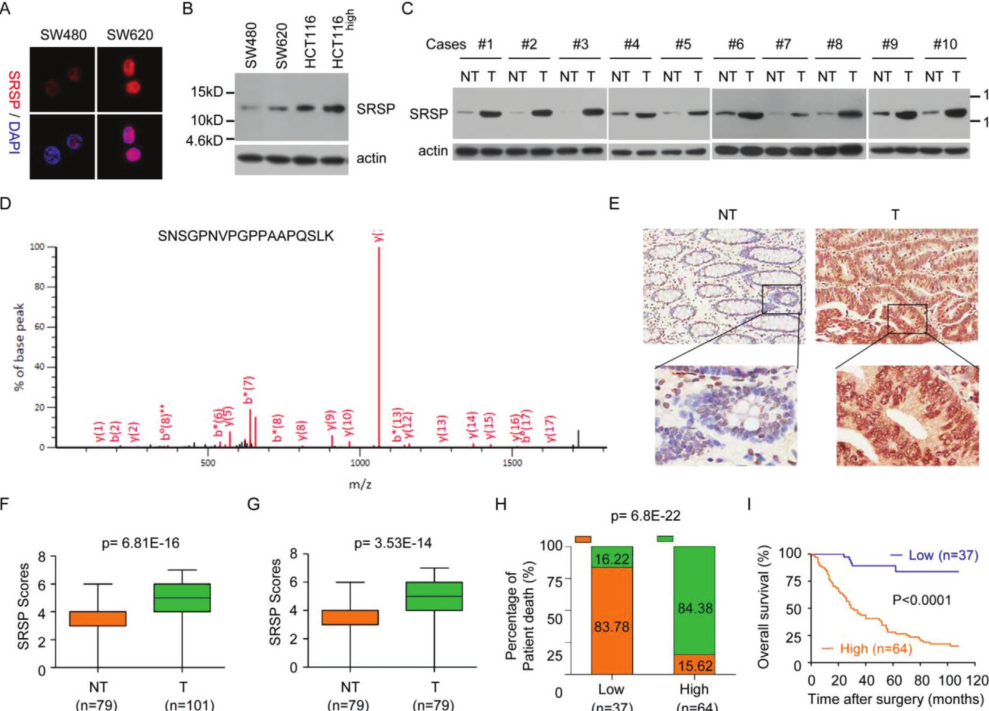
天然的内源性SRSP存在于结直肠癌,乳腺癌,卵巢癌和鼻咽癌癌细胞中,结肠癌和癌旁组织存在天然的内源性SRSP。质谱鉴定并验证了癌组织中天然内源产生的SRSP。综上所述,这些数据表明,SRSP是人类细胞和组织内源性自然产生的。IHC)分析了101个CRC组织样本和79个正常样本(包括79对CRC组织及其匹配组织)中的SRSP表达水平。CRC组织中SRSP表达水平显着高于相邻的癌旁组织。高SRSP水平的CRC患者比低SRSP水平的CRC癌症死亡风险更高。
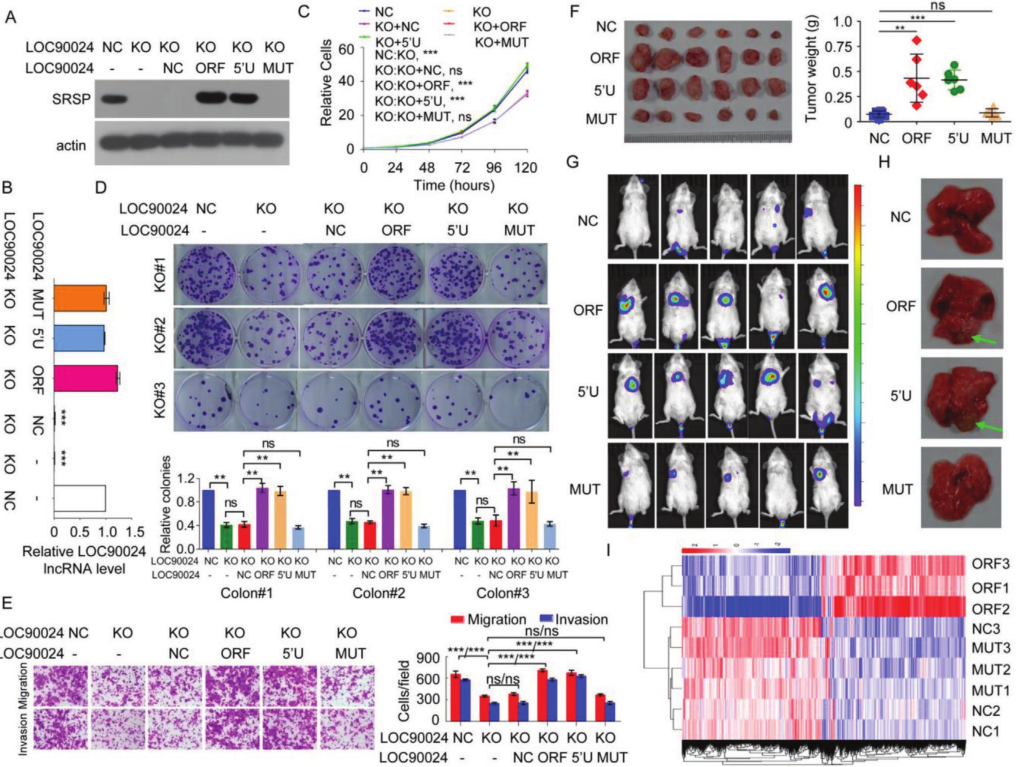
进一步研究LOC90024和SRSP在CRC肿瘤发生中的功能,使用CRISPR‐Cas9技术构建了LOC90024基因敲除(KO)细胞。LOC90024的 KO 可显着抑制癌细胞的增殖,集落形成,迁移和侵袭。在LOC90024 KO细胞恢复了lncRNA表达,LOC90024 ORF和5'UTR-ORF构建体可促进癌细胞增殖,集落形成,迁移和侵袭,而LOC90024 5'UTR-ORFmut构建体不会改变CRC细胞的增殖,集落形成,迁移和侵袭。ORFmut标志 5'UTR-ORFmut-Flag表达的体内异种移植肿瘤生长与空白载体表达相似。这些数据表明,SRSP而非LOC90024 lncRNA本身可促进肿瘤发生。
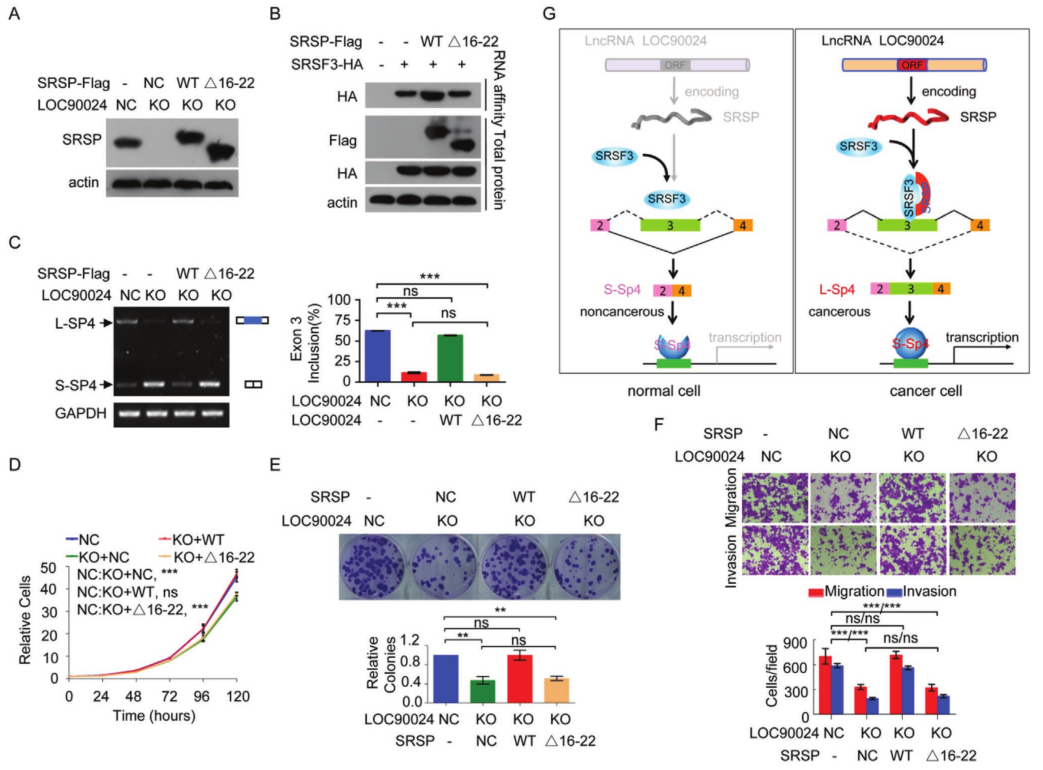
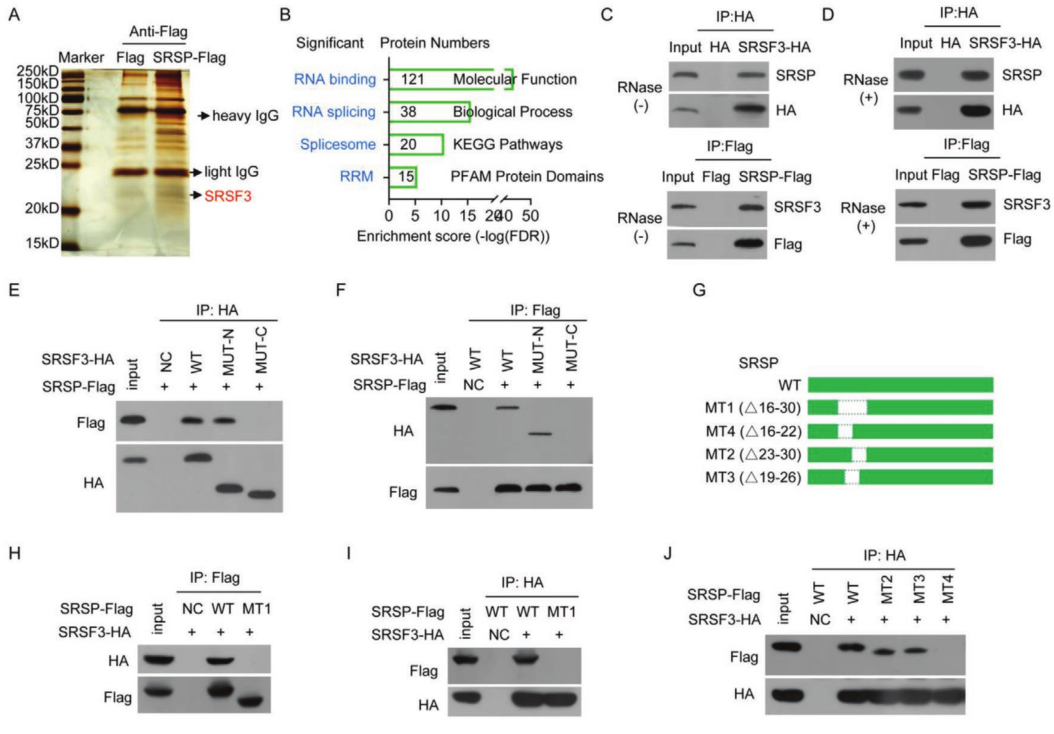
为了揭示SRSP促进肿瘤发生和癌症进展的机制,通过蛋白组学分析寻找了SRSP的相互作用蛋白。质谱鉴定出总共574种SRSP相互作用蛋白。GO分析显示,这些与SRSP相互作用的蛋白主要被归类为RNA结合蛋白,这表明SRSP可能主要通过与剪接调节剂相互作用来调节RNA剪接。SRSF3是一种RNA剪接因子和RNA结合蛋白。实验证实SRSP可以与SRSF3相互作用。进一步研究了SRSP是否通过RNA与SRSF3相互作用。在存在RNase处理的情况下,SRSP仍与SRSF3相互作用,表明SRSP与SRSF3的相互作用不依赖于RNA。
SRSF家族蛋白在N端包含一个RRM以与RNA结合,在C端包含一个RS结构域以与其他伴侣蛋白结合。将C端和N端突变在HEK293T细胞中共表达。只有包含N末端的SRSF3才能与SRSP相互作用。为了确定SRSP的哪些区域或残基与SRSF3相互作用,构建了一系列SRSP-Flag突变体,我们证明了SRSP(1–60 aa)的N末端与SRSF3相互作用。只有包含16-30个aa区域的SRSP才能与SRSF3相互作用,而该区域的脯氨酸-精氨酸-精氨酸(PRR)基序对于相互作用不是必需的,确认SRSP的16-22aa区域与SRSF3相互作用。4J)。因此,SRSP中的GQPCGSP氨基酸(16-22氨基酸区域)是其与SRSF3 N末端相互作用的原因。
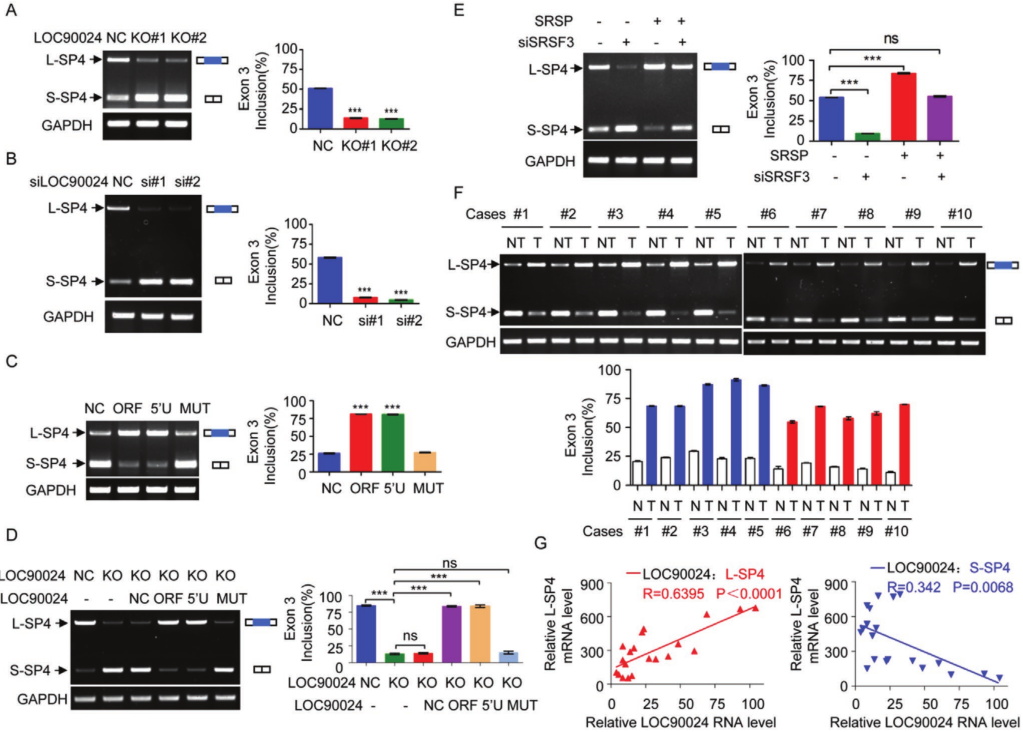
发现SRSP促进了TF Sp4外显子3的掺入,从而促进了长Sp4剪接变体形成(L-Sp4),并抑制了没有外显子3的短Sp4剪接变体形成(-S-Sp4)-。 当敲除SRSP表达时,有靶向RNA测序证实了Sp4外显子3的敲除。SRSF3结合位点的突变完全废除了SRSF3与Sp4 E3(848–866)RNA探针的结合。 但是,SRSP本身并未直接结合这些Sp4外显子3 RNA探针,这表明SRSP无法直接识别并结合Sp4外显子3。进一步研究了SRSP对SRSF3与Sp4外显子3结合的影响。 我们发现,当SRSF3结合位点在Sp4的外显子3中发生突变时,SRSP的过表达显着增强了SRSF3与Sp4的E3(848-866)的结合,但并未诱导SRSF3与Sp4的第4外显子结合。此外,SRSF3与Sp4外显子3的结合以SRSP剂量依赖性方式增加。

鉴于SRSP增强了SRSF3对Sp4外显子3的结合和识别,进一步研究了SRSP对Sp4 premRNA剪接的影响。 LOC90024的KO损害了Sp4外显子3的包含,从而减少了L‐Sp4的形成并增强了S‐Sp4的形成。在LOC90024-KD细胞中获得了相似的结果。 LOC90024 ORF和5′UTR‐ORF(5′U)(编码SRSP)的过表达增加了L-Sp4的形成并减少了S-Sp4的形成,而LOC90024 5′UTR‐ORFmut(MUT)的过表达并没有改变Sp4 premRNA的剪接。 我们用LOC90024 ORF,5'UTR-ORF(5'U)或5'UTR-ORFmut(MUT)载体在LOC90024 KO细胞中恢复了LOC90024的表达。 SRSP重新表达后,LOC90024 KO受损的Sp4外显子3的内含物恢复到对照水平,但LOC90024 lncRNA没有恢复。 此外,SRSP不会改变Sp4 mRNA的总水平。 SRSF3的KD减弱了SRSP过表达诱导的Sp4外显子3增加的包涵性。临床肿瘤组织样本中研究了Sp4 pre-mRNA剪接。 与匹配的相邻非肿瘤组织相比,肿瘤组织中的L‐Sp4 mRNA水平升高而S‐Sp4 mRNA水平降低。临床组织样品中L‐Sp4水平与SRSP水平呈正相关,而S‐Sp4水平与SRSP水平呈负相关。总体而言,SRSP促进了Sp4外显子3的SRSF3依赖性包涵,以诱导L-p4形成并抑制S-Sp4形成。
L-Sp4剪接变体编码784-aa蛋白L-Sp4亚型,而不带外显子3的S-Sp4剪接变体由于帧偏移而编码49-aa肽S-Sp4亚型。为了确定L-Sp4和S-Sp4在CRC肿瘤发生中的作用,我们恢复了Sp4-KD大肠癌细胞中抗Sp4 siRNA的L-Sp4或S-Sp4的表达。Sp4的KD可抑制HCT-116和SW480细胞的生长、集落形成、迁移和侵袭。在重新表达L-Sp4后,HCT-116和SW480 CRC细胞中Sp4的KD诱导的细胞生长、集落形成、迁移和侵袭的减少可以有效地恢复到控制水平,表明L-Sp4亚型具有致癌功能,而S-Sp4亚型没有致癌作用功能。

为了研究SRSP是否促进SRSF3与Sp4外显子3的结合、Sp4外显子3的包含以及通过与SRSF3的相互作用促进细胞的肿瘤发生,在LOC90024 KO细胞中恢复野生型SRSP或SRSF3结合缺陷SRSPΔ16–22突变体的表达。当SRSP中SRSP与SRSF3的结合区域被删除时,SRSP没有增加SRSF3与Sp4的E3(848–866)的结合。当SRSP中SRSP与SRSF3的结合区域被删除时,SRSP没有诱导Sp4第3外显子的包含以促进L-Sp4剪接变体的形成。从功能上讲,野生型SRSP的再表达有效地将LOC90024 KO诱导的细胞生长、菌落形成、迁移和侵袭的降低恢复到对照水平,而SRSF3结合缺陷SRSPΔ16–22突变体没有。因此,这些结果表明,SRSP主要通过与SRSF3的相互作用促进Sp4剪接和肿瘤发生。