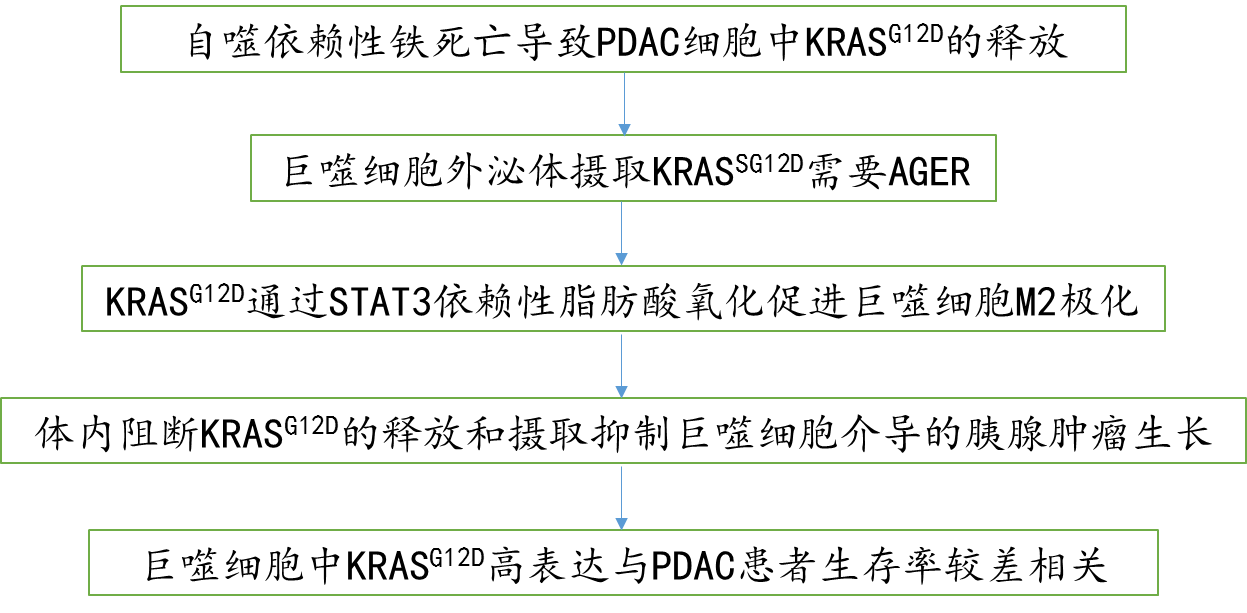
KRAS介导自噬依赖性铁死亡驱动肿瘤相关巨噬细胞的极化
研究背景:
KRAS是人体肿瘤中最常见的突变癌基因。尽管很多研究报道了KRAS突变对癌细胞的影响,但是致癌KRAS的激活对免疫细胞的直接影响仍不清楚。文章研究细胞外KRASG12D作为癌细胞-巨噬细胞间通信的关键中介(IF= 9.77)。
技术路线图:
研究结果:
1. 在自噬依赖性铁下垂癌细胞死亡中,KRASG12D在胞外释放
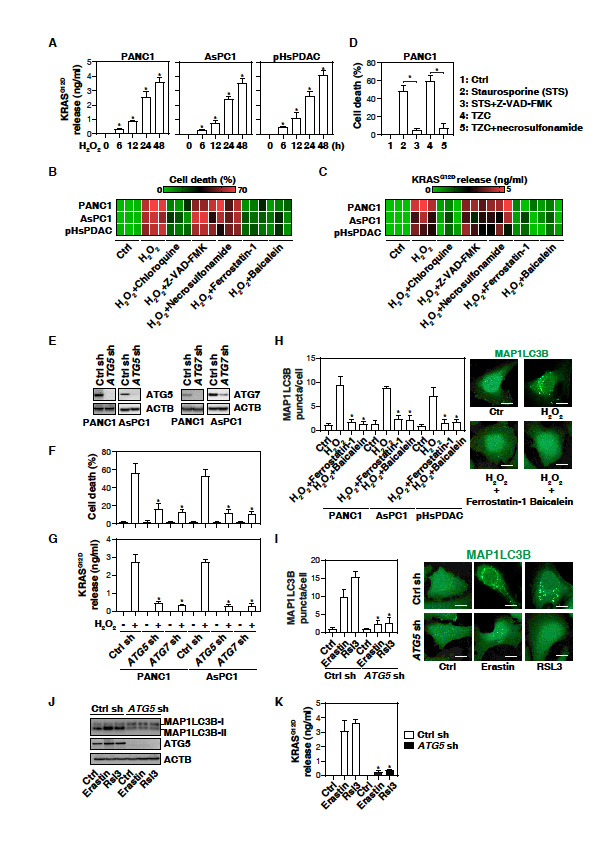
胰腺导管腺癌(PDAC)的发生和发展与氧化应激有关。在人PDAC细胞系(PANC1和AsPC1,都发生KRASG12D突变)确定氧化应激是否会诱导癌蛋白的释放。H2O2以时间依赖性的方式诱导NC1和AsPC1细胞以及人初级PDAC细胞(pHsPDAC)释放KRASG12D (图1A)。提示氧化应激引起PDAC细胞释放KRASG12D。H2O2可以触发多种形式的调控细胞死亡,比如细胞凋亡,坏死和自噬依赖的细胞死亡。H2O2诱导的细胞死亡(图1B)和PDAC细胞释放KRASG12D(图1C)被自噬抑制剂阻断(chloroquine),但是凋亡抑制剂或坏死凋亡抑制剂(Z-VAD-FMK和necrosulfonamide)没有这种作用。敲除自噬相关基因限制了H2O2诱导的细胞死亡和KRASG12D的释放(图1E-1G),进一步证实自噬依赖性细胞死亡促进氧化应激期间KRASG12D蛋白释放。
铁死亡抑制剂(ferrostatin-1和baicalein)阻断H2O2诱导的MAP1LC3B的斑点形成(图1H),抑制PDAC细胞死亡(图1B)和KRASG12D释放(图1C)。Erastin和RSL3等铁死亡诱导剂也能诱导MAP1LC3B斑点(图1I)、MAP1LC3B脂化(产生MAP1LC3B II)和KRASG12D释放(图1K)。敲除ATG5逆转这些效应(图1I - 1k)。结果提示铁死亡可导致PDAC细胞中KRASG12D蛋白的释放。
Figure1
2. 巨噬细胞外泌体摄取KRASSG12D需要AGER
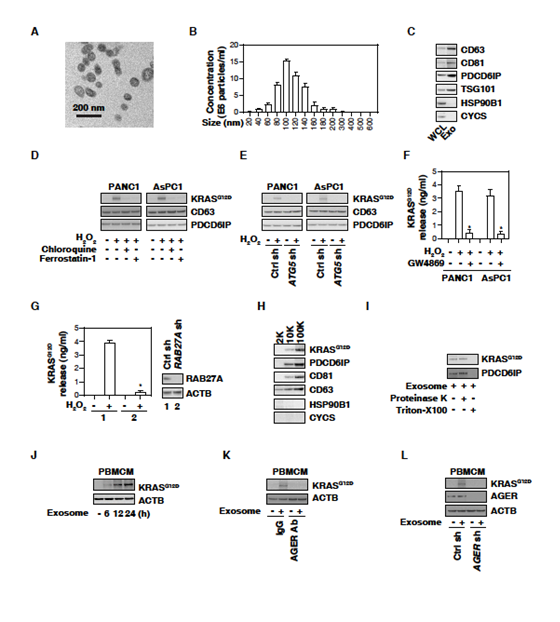
之后研究了KRASG12D蛋白是否存在于外泌体中。电子显微镜、纳米颗粒分析和WB检测外泌体分离方法的纯度。囊泡大小50-150nm (图2A、2B),WB检测CD63、CD81、PDCD6IP/ALIX、TSG101、HSP90B1 / GRP94和CYCS (图2C)进一步证明。在PANC1和AsPC1细胞分离的外泌体中,H2O2刺激导致KRASG12D蛋白(不影响CD63和PDCD6IP/ALIX)的增加;氯喹和ferrostatin-1阻断了KRASG12D蛋白的积累(图2D);敲除自噬相关基因ATG5或应用GW4869(外泌体生成和释放的抑制剂)抑制了H2O2诱导的KRASG12D蛋白的积累(图2E,F)。外泌体分泌的关键调节因子RAB27A的敲除也抑制了H2O2诱导的PANC1细胞释放KRASG12D (图2G)。KRASG12D和PDCD6IP/ALIX均对蛋白酶K不敏感,但被Triton X-100降解(图2I)。这些发现表明自噬依赖的铁死亡导致KRASG12D从外泌体释放。
之后研究肿瘤来源外泌体中的KRASG12D蛋白是否能被巨噬细胞吸收。人外周血单核细胞来源巨噬细胞(PBMCMs)通常不含KRASG12D,但以时间依赖性的方式吸收H2O2产生的癌症外泌体(图2J),提示肿瘤KRASG12D可进入巨噬细胞。AGER抗体抑制H2O2生成的癌症外泌体培养的PBMCMs对KRASG12D的摄取(图2K)。类似地,在PBMCMs中,敲除AGER损害了KRASG12D从肿瘤来源外泌体的摄取(图2L)。结果表明,AGER介导巨噬细胞摄取KRASG12D
Figure2
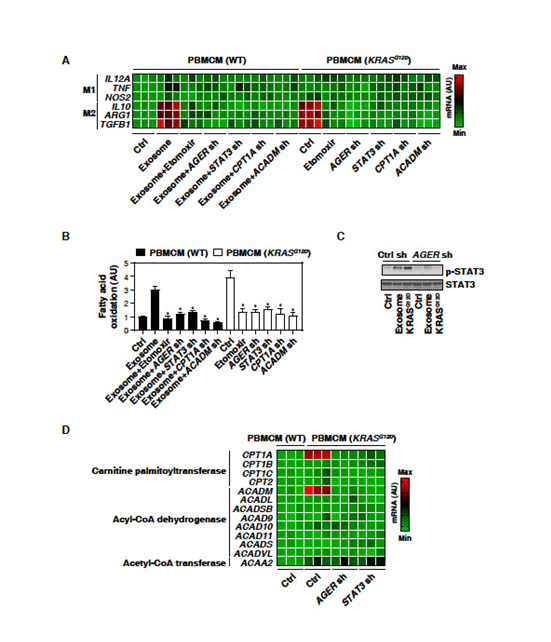
3. KRASG12D通过STAT3依赖性脂肪酸氧化促进巨噬细胞M2极化
巨噬细胞摄取KRASG12D可能影响巨噬细胞极化。敲除PBMCMs中KRASG12D,并检测M1分化(IL-12A、TNF和NOS2/iNOS)或M2极化(IL-10、ARG1和TGFB1)的生物标志物。
表达KRASG12D的PBMCMs中IL-10、ARG1和TGFB1 mRNA(M2极化)表达上调(M1极化不变);H2O2产生的癌症外泌体也增加了PBMCMs产生的M2 mRNA表达, AGER敲除所逆转(图3A)。因此,KRASG12D的摄取可能导致促癌的M2极化。脂肪酸氧化的情况类似(图3B)。脂肪酸氧化抑制剂Etomoxir降低了KRASG12D表达或PDAC细胞来源的外泌体的PBMCMs中IL10、ARG1和TGFB1 mRNA表达(图3A)。因此,脂肪酸氧化有助于KRASG12D介导的M2巨噬细胞极化。
为了解KRASG12D驱动的巨噬细胞中脂肪酸氧化加剧的机制,文章重点研究了STAT3。 WB分析发现在表达KRASG12D或肿瘤外泌体处理的PBMCMs中,STAT3的磷酸化水平上调(图3C)。与AGER的敲除类似,在KRASG12D或H2O2诱导的癌症外泌体的PBMCMs中,敲除STAT3减少脂肪酸氧化(图3B)和M2巨噬细胞极化(图3A)。因此,AGER介导的STAT3的活化需要KRASG12D-诱导脂肪酸氧化和巨噬细胞极化。
之后,确定参与脂肪酸氧化代谢途径的基因是否会被AGER介导的STAT3激活所调控。在表达KRASG12D的PBMCMs中,其他脂肪酸氧化相关基因(如CPT1B, CPT1C, CPT2, ACADL, ACADSB, ACAD9, ACAD10, ACAD11,ACADS,ACADVL,和ACAA2)上调 (图3 D)。这一效应被敲除AGER或STAT3抑制(图3D)。在KRASG12D表达或对癌症外泌体应答的PBMCMs中,敲除CPT1A和ACADM抑制脂肪酸氧化(图3B)和IL10、ARG1和TGFB1 mRNA表达(图3A)。结果表明AGER-STAT3通路对于KRASG12D诱导的脂肪酸氧化和随后的巨噬细胞M2极化至关重要。
Figure3
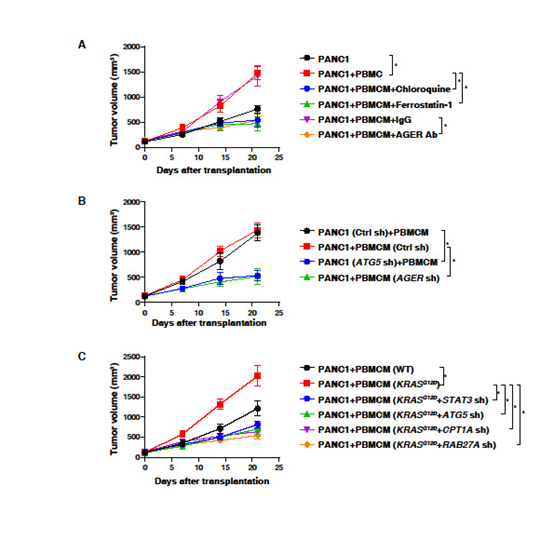
4. 体内阻断KRASG12D的释放和摄取抑制巨噬细胞介导的胰腺肿瘤生长
探索KRASG12D蛋白从癌细胞向巨噬细胞的可能路径。在NOD SCID小鼠背侧皮下注射PANC1细胞或PANC1细胞+PBMCMs。与单用PANC1细胞相比,PBMCMs促进了胰腺肿瘤的生长(图4A)。局部注射氯喹、ferrostatin-1或抗AGER抗体可抑制PANC1+PBMCMs引起的肿瘤的生长 (图4)。AGER的敲除(图4B)降低了由PANC1细胞+PBMCMs引起的肿瘤生长。此外,KRASG12D驱动的PBMCMs比野生型PBMCMs更能促进胰腺肿瘤的生长(图4C)。当STAT3、ATG5、CPT1A或RAB27A敲除时,PBMCMs表达KRASG12D的肿瘤加速作用消失(图4C)。这些发现表明,KRASG12D或KRASG12D转入PBMCMs可通过ATG5、STAT3、CPT1A和RAB27A等通路驱动PDAC的进展。
Figure4
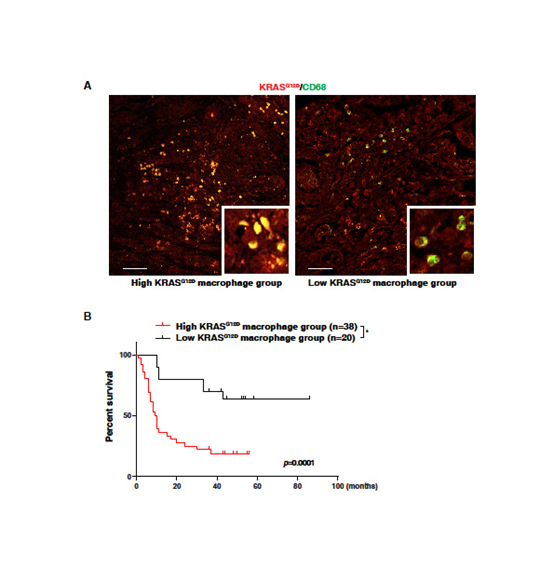
5. 巨噬细胞中KRASG12D的高表达与PDAC患者较差的生存相关
为了确定在人类胰腺癌中KRASG12D在巨噬细胞表达异常,分析59个PDAC病人(表1)。在CD68+巨噬细胞中检测出KRASG12D表达(图5A)。PDAC病人CD68+巨噬细胞中有高KRASG12D显示短生存期,比病人含有低KRASG12D在肿瘤浸润性CD68 +巨噬细胞 (图5B)。这些发现表明巨噬细胞摄取KRASG12D的增加可能促进了人PDAC的进展。
Figure5