






国自然热点-自噬
2020年国自然已尘埃落定,2021年国自然申请又即将开始。那么接下来小编给大家介绍一下国自然热点之一—自噬。自噬从2010到2017申请的相关项目一直在上升,近两年稍有回落,但也多达500多项。其中肿瘤学相关的研究遥遥领先。话不多说,今天和大家分享一篇自噬相关的文章。


这篇发表于影响因子10.717的“Cell Death & Differentiation”杂志的文章“DAPK3 inhibits gastric cancer progression via activation of ULK1-dependent autophagy” 报道了DAPK3在胃癌细胞中诱导自噬的抑癌作用。DAPK3在GC中的抑癌作用与自噬相关,DAPK3是一种新型的自噬调节因子,可以直接磷酸化ULK1并激活ULK1。因此,DAPK3可能是一个潜在的预后的自噬相关标志物。
结果:
1)DAPK3在胃癌细胞中显示肿瘤抑制活性
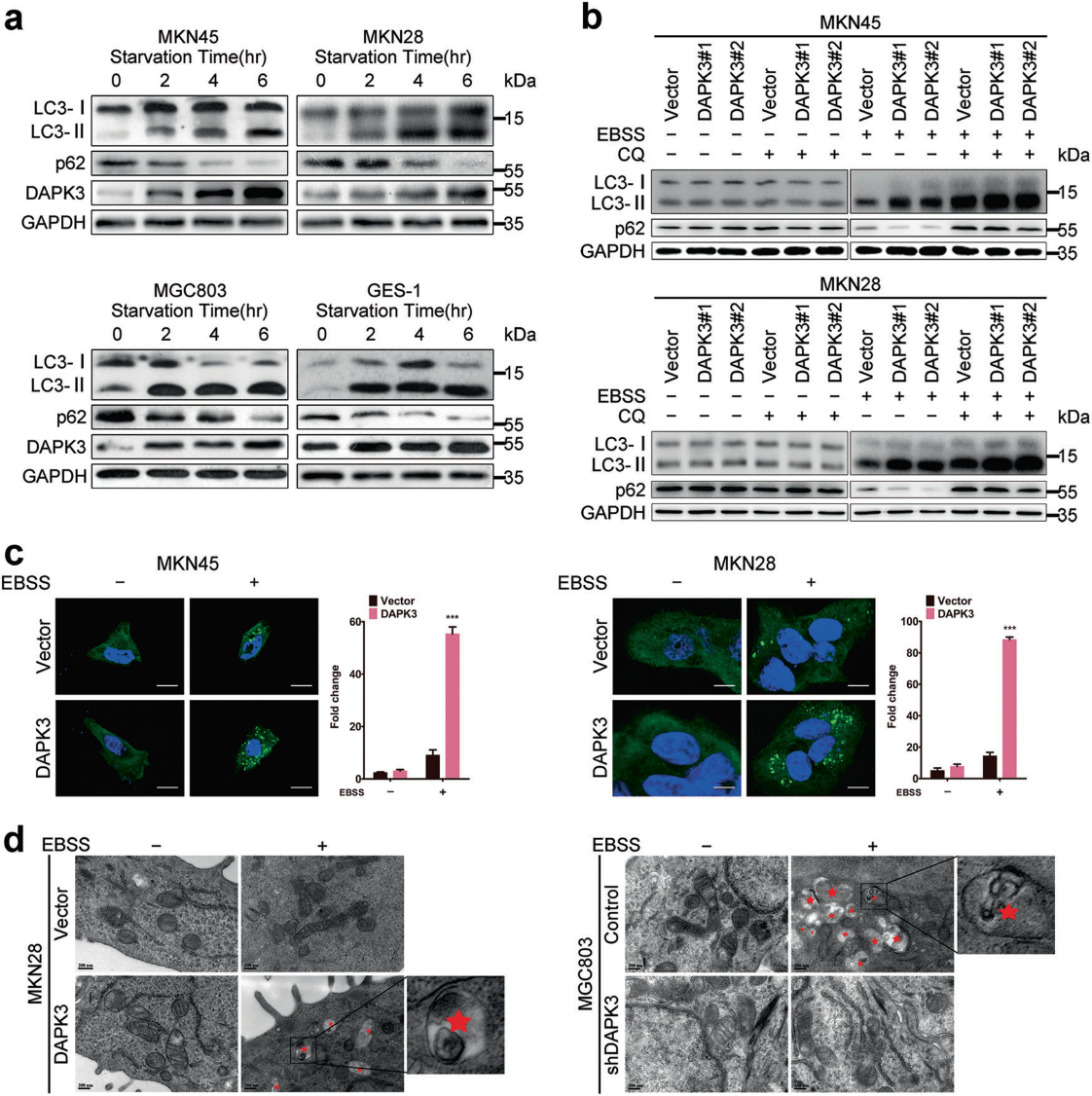
我们前期研究表明DAPK3的表达与胃癌晚期临床分期和不良预后呈负相关。为了深入了解DAPK3在胃癌细胞中的肿瘤抑制功能,我们进行了以下实验。 XTT增殖试验显示DAPK3显著抑制肿瘤细胞生长速率(图1a)。病灶形成和软琼脂试验显示DAPK3显著降低了依赖锚定和独立细胞中的集落频率和大小(图1b,c)。MGC803和GES-1细胞中DAPK3的敲除强烈促进细胞生长和集落形成(图1a-c)。为了验证DAPK3对体内肿瘤生长的影响,建立了皮下异种移植瘤小鼠模型。我们发现,空载体转染的MKN45细胞在注射后7天内就可以看到肿瘤,而DAPK3转染的MKN45细胞直到注射后3周才观察到肿瘤(图1d)。动力学图显示由DAPK3沉默的MGC803细胞产生的肿瘤明显大于来自对照细胞的肿瘤(图1d)。伤口愈合和transwell试验表明,DAPK3过表达后细胞迁移和侵袭明显减少,DAPK3敲除后细胞迁移和侵袭增加(图1e,f)。另外,与对照细胞相比,DAPK3过表达的MKN45和MKN28细胞显示出G0/G1期细胞百分比的显著增加。同时,当DAPK3沉默时,我们观察到MGC803和GES-1细胞中G0/G1期细胞的百分比显著降低(图1g)。

2)氨基酸剥夺诱导自噬需要DAPK3
我们研究了DAPK3是否调节胃癌细胞的自噬。蛋白质印迹分析显示,EBSS处理促进自噬后,内源性DAPK3蛋白水平显著增加(图2a)。类似地,DAPK3过表达增加了LC3-II/LC3-I比率。同时,在氨基酸饥饿条件下,SQSTM1/p62水平在DAPK3过表达的细胞中显著降低 (图2b)。此外,在DAPK3过表达的细胞中发现LC3的数量增加,表明DAPK3增强了自噬体的形成(图2c)。透射电镜显示,与对照细胞相比,DAPK3过表达细胞中含有细胞质结构和残留消化物质的自噬空泡显著增加(图2d)。MGC803和GES-1细胞中DAPK3的敲除显著损害了LC3-I向LC3-II的转化、SQSTM1/p62的降解、GFP-LC3的积累和氨基酸饥饿期间自噬空泡的形成(图2d)。因此,DAPK3是诱导氨基酸饥饿介导的自噬所必需的。
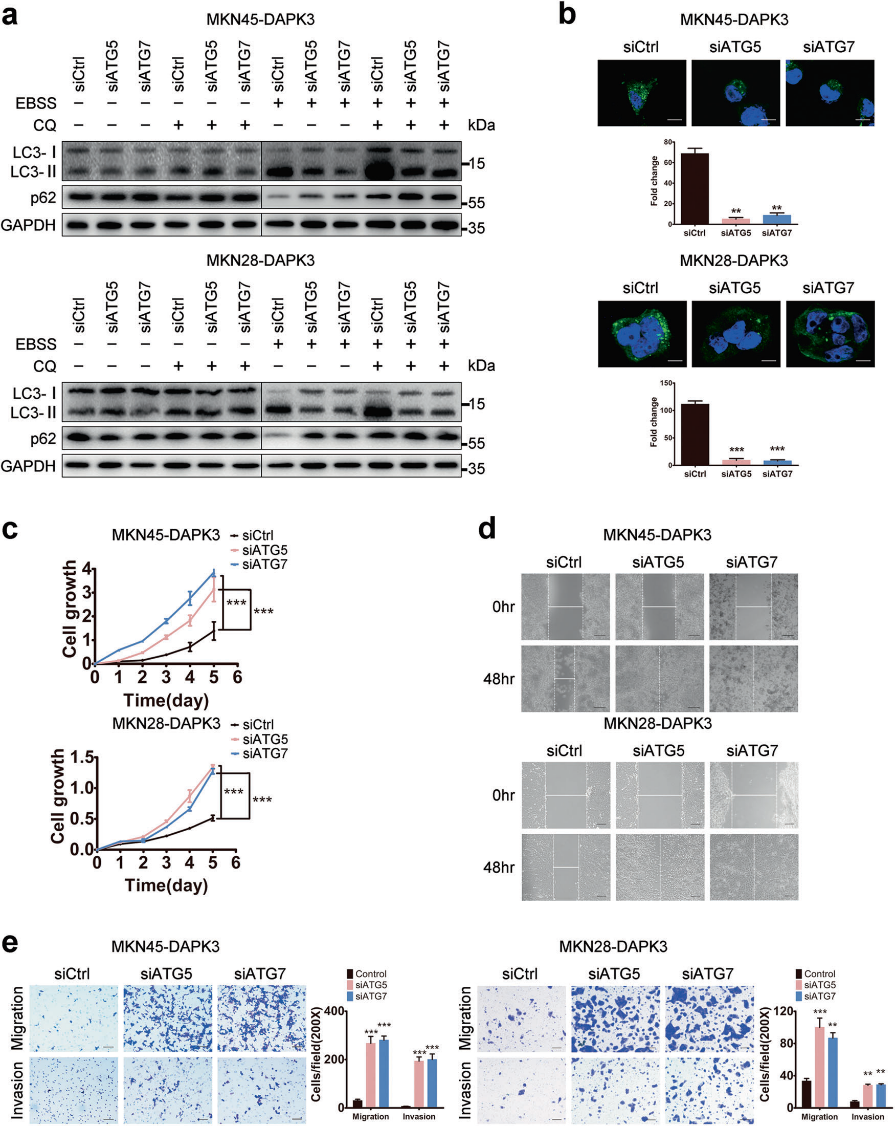
3) ATG5或ATG7缺失抑制的自噬会削弱DAPK3的肿瘤抑制功能
为了确定DAPK3介导的自噬是否与其肿瘤抑制功能相关,在DAPK3过表达细胞和空载体转染细胞中,抑制ATG5或ATG7。ATG5或ATG7的敲除显著降低了DAPK3过表达细胞中LC3-I向LC3-II的转化、SQSTM1/P62降解和GFP-LC3形成,表明DAPK3诱导的自噬受到ATG5或ATG7耗竭的损害(图3a,b)。接下来,我们研究了自噬抑制对DAPK3过表达细胞和空载体转染细胞的细胞增殖、迁移和侵袭的影响。细胞生长试验显示细胞生长速率在ATG5或ATG7缺失的DAPK3过表达细胞明显高于对照组细胞。同时,ATG5或ATG7敲除促进细胞迁移和侵袭(图3c-e)。这些结果表明,ATG5或ATG7敲除可以挽救DAPK3诱导的GC细胞的生长和转移阻滞。
4) DAPK3通过ULK1激活调节自噬
为了阐明DAPK3介导的自噬的分子机制,我们使用基于质谱的定量磷酸化蛋白质组分析来分析在氨基酸饥饿下DAPK3过表达的MKN28细胞和对照细胞中差异表达的蛋白质。与对照细胞相比,我们观察到DAPK3过表达细胞中Ser556的ULK1磷酸化增加了大约两倍。免疫印迹分析显示氨基酸饥饿后DAPK3过表达细胞中Ser556上ULK1磷酸化水平的显著上调(图4a)。我们还检测了Beclin-1的磷酸化状态,Beklin-1是ULK1的底物,是VPS34复合物激活和完全自噬诱导所必需的。正如预期的那样,DAPK3增加了Ser15的Beclin-1磷酸化,表明DAPK3调节ULK1激活(图4a)。此外,ULK1敲除强烈抑制自噬通量、自噬体形成,并增强DAPK3过表达细胞在体外的致瘤性(图4b-g)。在接种了ULK1沉默的MKN45-DAPK3细胞的裸鼠中,肿瘤大小显著增加,表明DAPK3介导的肿瘤抑制依赖于ULK1的激活(图4h)。

5) DAPK3通过Ser556的直接磷酸化激活ULK1
为了研究DAPK3直接磷酸化ULK1的可能性,我们使用了GPS软件来预测DAPK3的ULK1磷酸化位点,并发现ULK1可能在Ser556被DAPK3磷酸化(图5a)。DAPK3在Ser556磷酸化重组ULK1,提示DAPK3可以直接在Ser556磷酸化ULK1(图5b)。DAPK3还在体外激酶测定中强烈磷酸化Ser556 位点的ULK1,在氨基酸饥饿状态下,ULK1的Ser556位点磷酸化水平进一步提高(图5c)。然而,Ser556突变显著阻碍了该位点的磷酸化(图5c)。为了进一步验证体内ULK1中DAPK3磷酸化位点,我们通过免疫沉淀来检测Ser556位点ULK1磷酸化。正如预期的那样,在DAPK3过表达的细胞中观察到高水平的ULK1 Ser556磷酸化(图5d)。WT DAPK3过表达导致的Ser556 ULK 1磷酸化的增加明显高于DAPK3 K42A过表达导致的增加,表明DAPK3激酶活性是ULK1 Ser556磷酸化所必需的(图5e)。
接下来,我们将HA-ULK1作为关键分子进行免疫沉淀,并分析了在DAPK3过表达细胞和空载体对照细胞中ULK1和其他ULK1复合物成分之间的相互作用。DAPK3表达在饥饿条件下显著增强了WT ULK1与其结合配偶体FIP200、ATG13和ATG101之间的相互作用,而ULK1中Ser556磷酸化位点的缺失部分削弱了这些相互作用(图5d)。ATG101在anti-Flag免疫沉淀中被发现,饥饿后WT DAPK3细胞中ATG101的量增加。饥饿时,FIP200和ATG13与标记的WT DAPK3共免疫沉淀(图5e)。ULK1通过直接磷酸化Beclin1和ATG14L,有利于VPS34复合物的激活。于是我们通过蛋白质印迹分析探讨了DAPK3在VPS34复合物中的作用。研究表明,在饥饿条件下激活HEK293T细胞中的VPS34复合物需要ULK1 Ser556的磷酸化和DAPK3激酶活性(图5f,g)。总的来说,这些数据表明DAPK3对ULK1的磷酸化增强了ULK1复合物的形成和饥饿时VPS34复合物的活化。

6) ULK1中的Ser556磷酸化促进DAPK3介导的自噬和肿瘤抑制
为了确定ULK1磷酸化在DAPK3调节的自噬和肿瘤抑制中的功能意义,我们将WT ULK1或S556A ULK1 cDNA转染到DAPK3过表达和空载体的MKN45细胞中。DAPK3过表达的MKN45细胞在shRNA诱导的内源性ULK1下调下表现出自噬缺陷和致瘤性增加。WT ULK1 cDNA的重建恢复了自噬和肿瘤抑制(图6a,c,d)。为了进一步评估UKL1磷酸化对体内肿瘤生长的影响,我们进行了肿瘤异种移植实验。带有WT ULK1的DAPK3过表达MKN45细胞的异种移植物生长速度比来自S556A突变表达细胞的异种移植物生长速度慢。与空载体对照细胞相比,WT ULK1表达在DAPK 3过表达的MKN45细胞中显示出更显著的肿瘤生长抑制作用(图6g)。然后我们分析了DAPK3激酶活性对自噬诱导和肿瘤抑制的重要性。与DAPK3 K42A细胞相比,含有DAPK3 WT的MGC803细胞显示出更高的自噬水平和更低的细胞生长率、运动性和侵袭性(图6b,e,f)。综上所述, DAPK3的激酶活性和Ser556的ULK1磷酸化是DAPK3功能所必需的。

7) DAPK3的表达与手术GC标本中Ser556和LC3B的ULK1磷酸化呈正相关
我们的体外研究表明,DAPK3对ULK1 Ser556的磷酸化对自噬诱导和肿瘤抑制至关重要。相关分析表明DAPK3表达与GC组织中pULK1 Ser556和LC3B的免疫染色呈正相关(图7a,b)。Kaplan-Meier分析显示,DAPK3和pULK1 Ser556共表达的患者具有更长的总生存期和癌症特异性生存期。同样,DAPK3和LC3B的共同表达预示着患者的良好预后(图7c,d)。这些数据表明DAPK3相关的自噬对人胃癌的进展有明显的抑制作用。
