国自然热点之焦亡
细胞焦亡(Pyroptosis)是一种炎症细胞程序性死亡方式,主要通过炎症小体介导包含半胱天冬酶-1(Caspase-1)在内的多种Caspase的激活,造成包括GSDMD在内的多种Gasdermin家族成员发生剪切和多聚化,造成细胞穿孔,进而引起细胞死亡。近些年细胞焦亡的中标数一直在上升,特别是2018和2019,这两年是呈现直线上升。接下来,我们通过一篇文献来了解焦亡研究的套路。

这篇题为“Neural progenitor cell pyroptosis contributes to Zika virus-induced brain atrophy and represents a therapeutic target”的文章讲述了ZIKV破坏神经发生并导致小头畸形的机制。在本研究中,我们发现ZIKV通过诱导caspase-1和 GSDMD介导的焦亡直接影响神经祖细胞的发育,将ZIKV感染与小头畸形的发生联系起来。重要的是,caspase- 1缺失或其抑制剂VX-765治疗可减少ZIKV诱导的炎症反应和焦亡,显著减轻体内神经病理学和脑萎缩。
结果:
1) ZIKV在新生鼠脑中复制并导致严重的神经病理学改变和脑萎缩
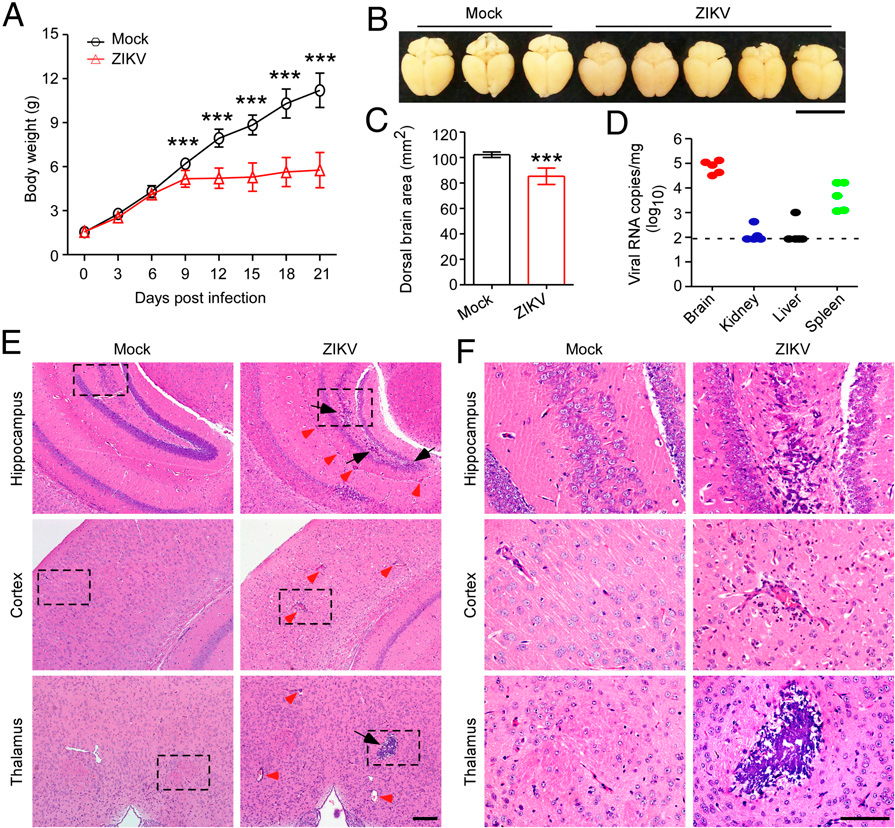
为了评估ZIKV感染在小鼠脑发育过程中是否具有致病性,WT幼鼠皮下感染5×105个ZIKV PFU。值得注意的是,在从感染后3dpi到21 dpi期间,ZIKV感染的小鼠显示出比模拟感染的小鼠更低的体重(图1A)。此外,在ZIKV感染的小鼠中有明显的脑萎缩,表现为在21 dpi时背侧脑面积减少(图1 B和1C)。使用qRT-PCR,证实在ZIKV感染小鼠的大脑中存在大量病毒核糖核酸拷贝,表明观察到的脑萎缩与ZIKV感染密切相关(图1D)。为了进一步研究ZIKV感染对脑发育的可能致病作用,对小鼠脑的组织切片进行了神经病理学检查。值得注意的是,ZIKV感染的小鼠大脑显示出大量中性粒细胞浸润、坏死和主要在海马、丘脑和皮层区域的正常细胞结构的破坏,表明大脑中存在严重的局灶性炎症,而模拟感染不会引起病理变化(图1 E和F)。此外,ZIKV感染小鼠的大脑经常出现单核细胞和小胶质细胞的血管周围套袖。综上所述,我们的发现表明ZIKV感染导致脑萎缩,并在所用的小鼠感染模型中诱导强烈的炎性脑损伤。
2) ZIKV感染导致体内NPC焦亡
基于我们前述对寨卡病毒感染小鼠大脑炎症损伤的观察,我们进一步探索ZIKV感染是否也能在NPC人群的大脑中引发caspase-1介导的焦亡。如图2 A和图2B所示,当在具有指示标记的NPC小生境中对裂解的caspase-1 (p10)和PI进行染色时,感染寨卡病毒的小鼠显示出丰富的裂解的caspase-1和PI+细胞。
由于焦亡与炎症小体激活有联系,接下来在从ZIKV感染和模拟感染小鼠获得的脑组织标本中评估了更广泛的炎症小体基因。如图2C所示,在小鼠脑组织标本中可检测到所有与炎症体相关的基因,与模拟感染相比,寨卡病毒感染的小鼠中IL1B、IL-18、CASP1、ASC和GSDMD转录物的水平显著升高。此外,与模拟对照相比,NLRP3在ZIKV感染的脑中的表达显著增加 (图2D)。在ZIKV感染的大脑中,caspase-1和GSDMD的裂解程度都很高(图2E)。综上所述,这些数据支持了ZIKV感染NPCs并在体内引发焦亡的观点。

3) ZIKV在体外感染人NPCs并诱导焦亡
为了进一步了解ZIKV和神经发生障碍之间的可能联系,我们接下来评估了ZIKV诱导的人NPCs(hNPCs)的焦亡的生物学效应。在ZIKV感染的hNPCs中,出现典型的炎性改变,包括细胞肿胀和膜破裂(图3A)。hNPCs中的ZIKV感染通过细胞中ZIKV E抗原和SOX2的共同存在被证实(图3B)。此外,通过透射电子显微镜(TEM)进行的超微结构分析也揭示了坏死的形态学变化,显示了ZIKV感染后染色质聚集、线粒体肿胀和质膜破裂(图3C)。我们接下来检查了细胞裂解物中caspase-1的活性,数据显示受ZIKV感染的细胞中caspase-1活性增加(图3D)。此外,用PI/Hoechst 33342双染色法进一步验证了炎症坏死细胞,正如所料,感染ZIKV的hNPCs发生了焦亡(图3 E和F)。此外,与模拟感染相比,ZIKV感染显著增加了乳酸脱氢酶的释放量,而与siRNA阴性对照组相比,沉默caspase-1的表达减少了ZIKV感染引起的乳酸脱氢酶的释放量(图3G),这表明caspase-1介导的NPCs的焦亡有助于ZIKV发病机制。与模拟感染的对应细胞相比,ZIKV感染触发了hNPCs中caspase-1和GSDMD的裂解(图3H)。 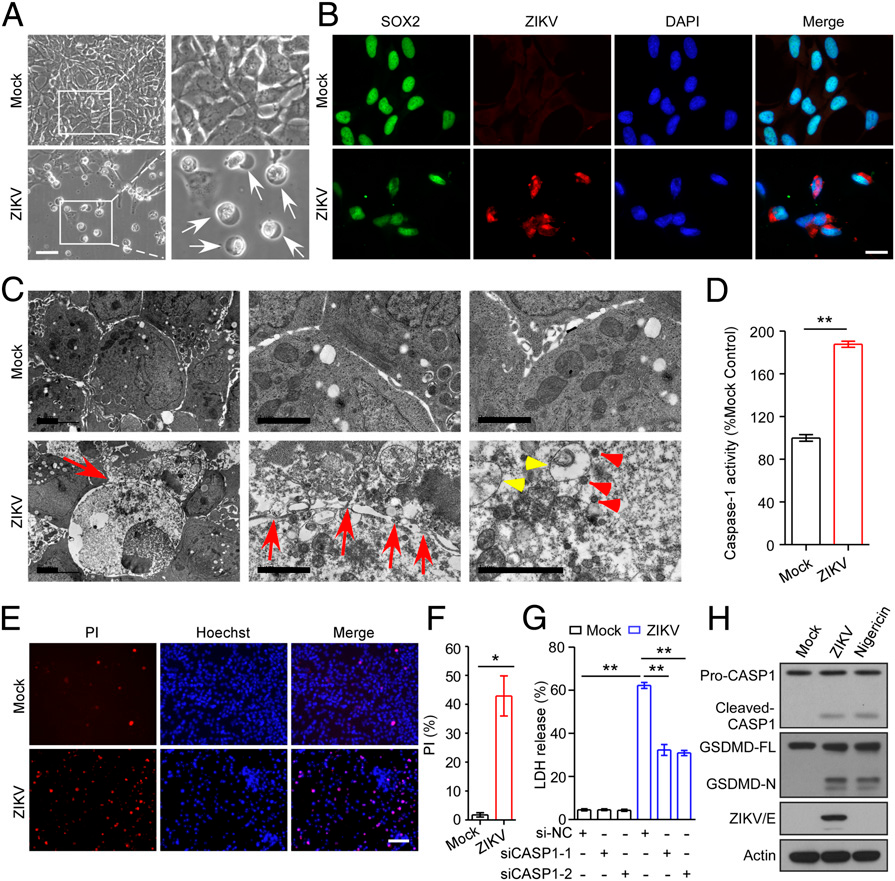
4) ZIKV诱导人类神经球的焦亡
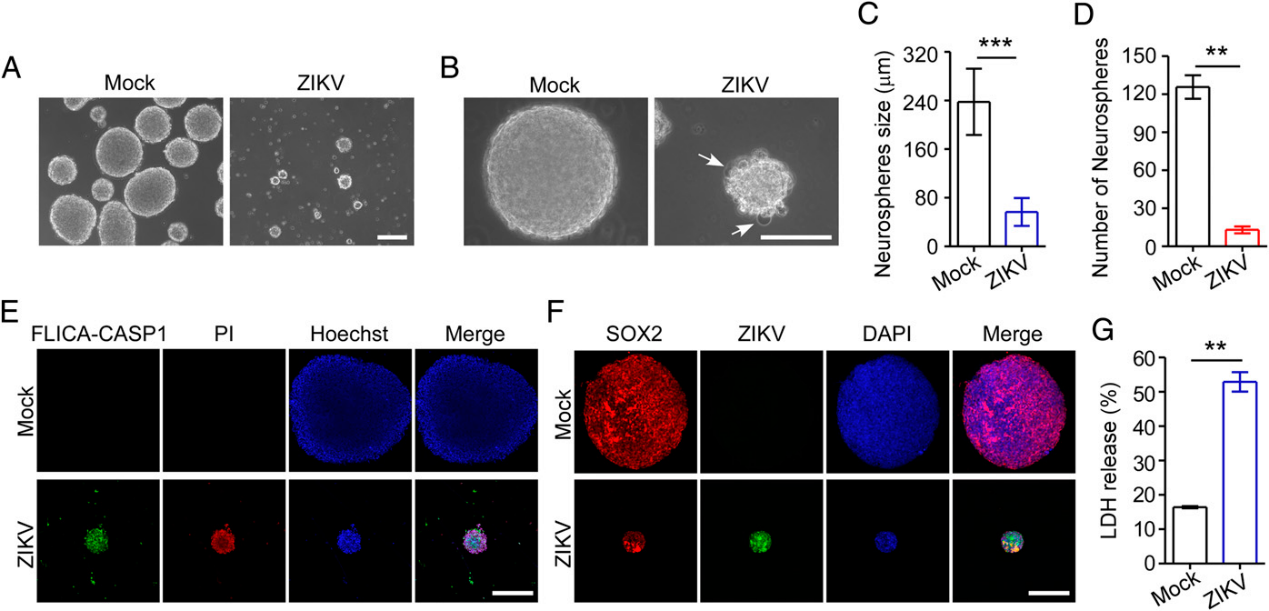
为了进一步说明神经发生过程中ZIKV感染的生物学效应,我们接下来用ZIKV感染神经细胞的3D培养物,然后观察神经球的形成。我们发现模拟感染的hNPCs产生圆形神经球,而ZIKV感染的神经球表现出形态学异常,并有焦亡的迹象(图4 A和4B)。此外,模拟感染的神经球随着时间的推移继续生长,但只有少数ZIKV感染的神经球以较小的尺寸存活(图4A-D)。为了进一步证实感染ZIKV的神经球发生了焦亡,我们用活性caspase-1和PI对神经球进行了双重染色。如图4E所示,感染ZIKV的神经球含有染色强烈的活性caspase-1和PI,这表明ZIKV感染可以诱导神经球的焦亡。神经球感染ZIKV通过神经球中存在ZIKV E抗原和SOX2被证实(图4F),此外,还测定了ZIKV感染后神经球的乳酸脱氢酶释放,表明ZIKV感染诱导了神经球的乳酸脱氢酶释放(图4G)。
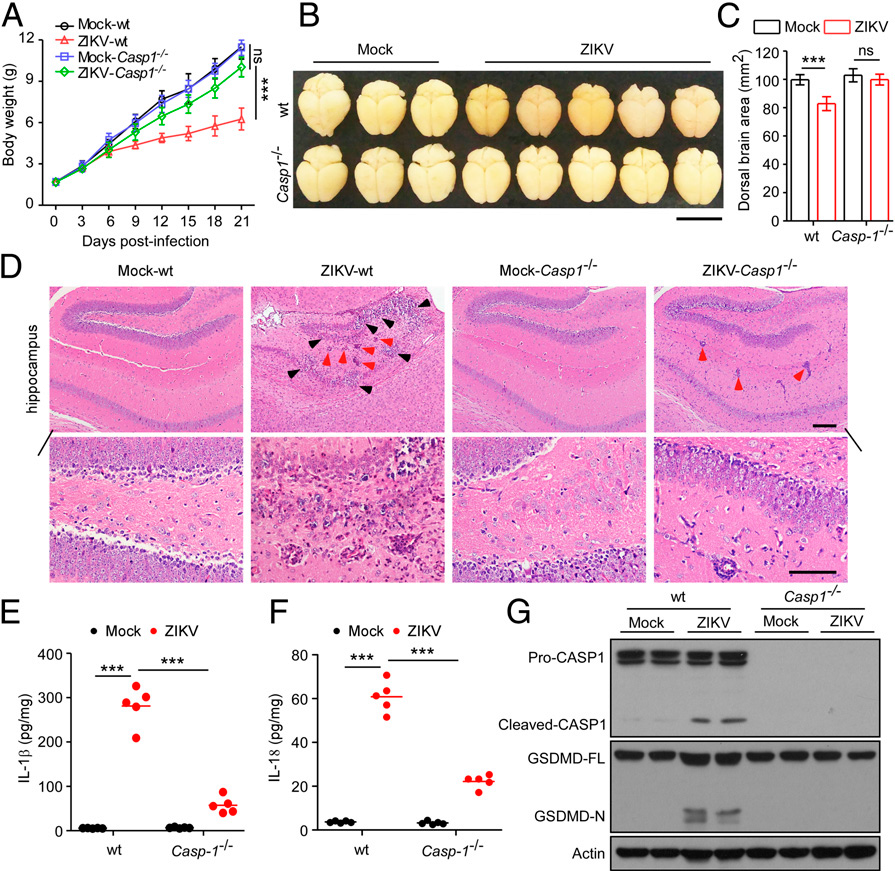
5) Caspase-1的缺失逆转ZIKV诱导的神经病理学改变和脑萎缩
我们生成了caspase-1-null小鼠,以进一步评估焦亡ZIKV诱导的脑畸形发病机制中的作用。我们发现与模拟感染组相比,感染ZIKV的WT小鼠体重较低,而ZIKV感染的Casp1−/−小鼠体重与模拟感染组在21日龄时的体重相似(图5A)。与ZIKV感染的野生型小鼠相比,ZIKV感染的Casp1−/−小鼠的严重脑萎缩被彻底消除(图5 B和5C)。组织病理学检查进一步表明,与ZIKV感染的野生型小鼠相比,Casp1−/−小鼠都免于炎症诱导的损伤(图5D)。此外,当在脑组织中测量促炎细胞因子IL-1β和IL-18的表达水平时,我们发现与野生型小鼠相比,在感染ZIKV的小鼠中,caspase-1的减少消除了IL-1β和IL-18的增加(图5 E和F)。此外,我们的结果还表明,ZIKV感染引发了GSDMD的裂解,而caspase-1缺失减弱了GSDMD的裂解(图5G)。因此,由ZIKV引起的神经病理学和脑萎缩可以通过废除caspase-1介导的焦亡来抑制。
6) VX-765治疗减轻ZIKV感染小鼠的神经病理学和脑萎缩
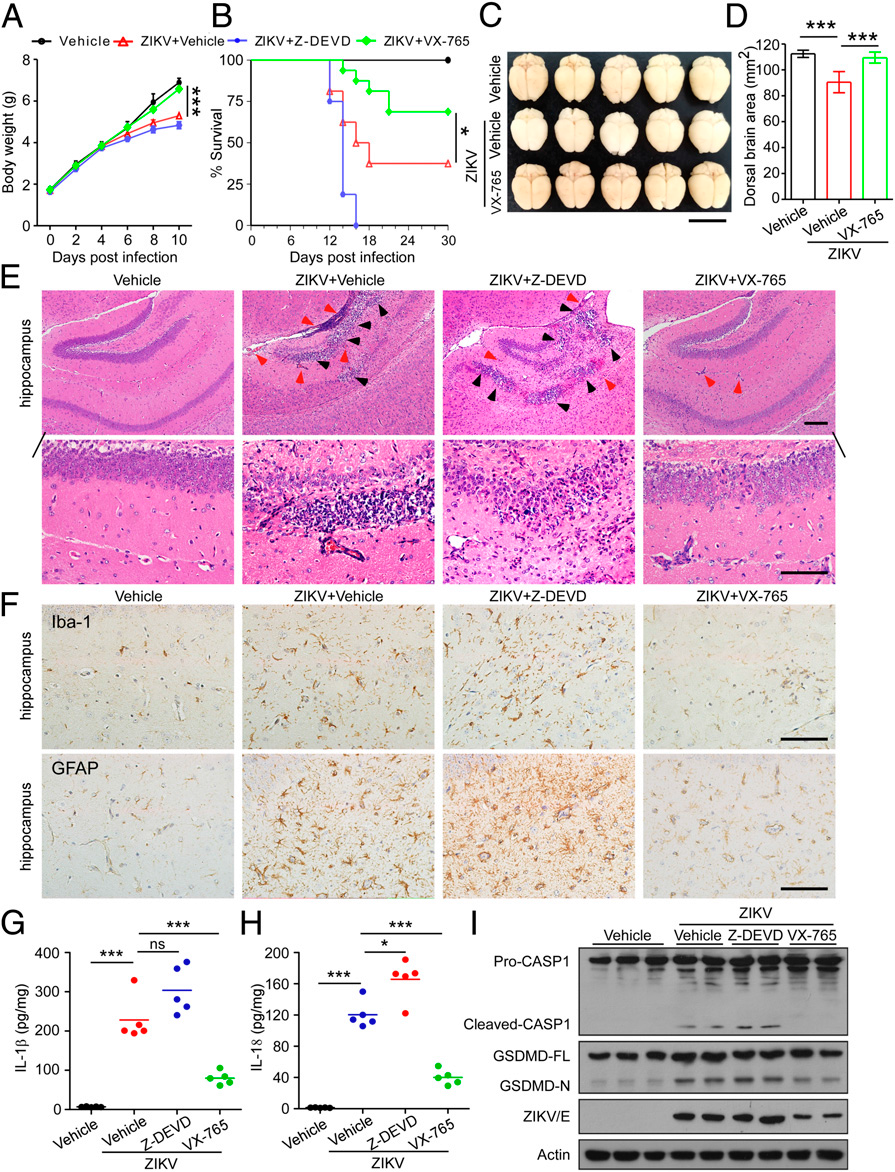
caspase-1在介导ZIKV相关神经病理学中的重要作用促使我们研究caspase-1是否可以成为神经病理学和小头畸形的潜在治疗靶点。VX-765是是一种强效和选择性的caspase-1抑制剂。结果表明,对照载体或Z-DEVD-FMK处理的小鼠比模拟感染的对照小鼠增重慢,而VX-765处理的小鼠没有出现这种结果(图6A)。30天后,ZIKV感染小鼠的存活率仅为38%,相比之下,VX-765治疗的小鼠的存活率更高,为69%。尽管用Z-DEDFMK治疗的所有小鼠在感染后第16天死亡(图6B),但进一步的观察表明,Z-DEVD-FMK治疗的小鼠与用载体或VX-765治疗的小鼠在没有ZIKV感染的情况下在体重变化和存活率方面没有显著差异。此外,与载体对照处理的ZIKV感染小鼠相比,VX-765处理减轻了由ZIKV感染引起的严重脑萎缩(图6 C和6D)。
进一步的组织病理学检查显示,在ZIKV感染的对照组和Z-DEVD-FMK治疗的小鼠的海马中观察到大量的中性粒细胞浸润和坏死位点,但在VX-765治疗后显著减弱(图6E)。免疫组织化学(IHC)对小胶质细胞和星形胶质细胞的Iba-1和GFAP特异性标记物的分析分别用于评估大脑中的神经炎症。如图6F所示,与模拟感染小鼠相比,ZIKV感染对照组和Z-DEVD-FMK治疗组小鼠的海马体中的Iba-1和GFAP水平显著更高。值得注意的是,VX-765的治疗降低了Iba-1和GFAP的表达水平,与未感染小鼠的大脑相当,这表明VX-765能够逆转神经炎症。
此外,在暴露于ZIKV的对照载体处理的小鼠的脑组织中,促炎细胞因子IL-1β和IL-18的表达水平上调,而这种上调被VX-765处理抑制,但不被Z-DEVD-FMK抑制(图6 G和H)。一致地,在VX-765治疗后,ZIKV感染小鼠的大脑中caspase-1和GSDMD的蛋白酶裂解受到抑制,这表明VX-765抑制了体内与ZIKV感染相关的焦亡(图6I)。