






癌症的劲敌——BORIS在DNA损伤修复中的新角色
印迹位点调节剂( BORIS )的类似物在大多数癌症中都有表达,往往与患者的短暂生存和不良预后有关。然而,其作用机制尚未阐明,也没有已知的BORIS抑制剂。本研究坚定了一种BORIS抑制剂,强调了BORIS中ADP核糖基化的重要性,揭示了BORIS在DNA损伤修复中的新功能。本研究于2022年8月发表在《Molecular Cancer》IF:41.444期刊上。
技术路线:

主要研究结果:
1. BORIS - binding肽的特征和选择
BORIS-N1 - 258和BORIS-del N1 - 258通过交替洗脱来自Ph.D.™-12噬菌体展示肽库中富集BORIS-N1 - 258结合的噬菌体克隆。BORIS-N1 – 258用来除去非特异性的噬菌体克隆,BORIS-del N1 – 258用来随后的噬菌体克隆。经过四轮富集,洗脱的噬菌体滴度达到一定程度。然后,用BORIS-N1-258涂层板ELISA法对这9种多肽的富集噬菌体克隆进行检测(图1C)。噬菌体克隆9对BORIS-N1 - 258蛋白的结合亲和力最高。
利用Fortebio Octet RED系统,合成多个克隆(# 9和# 2)中发现或具有高结合亲和力( # 9 )的展示肽,通过生物层干涉技术( BLI )分析其与BORIS-N1 - 258蛋白的物理相互作用。BORIS-N1 - 258蛋白与生物素结合并负载到SSA传感器上。合成的多肽用0.02 % Tween20溶解于PBS中。肽9 (肽序列为VHWDFRQWWQPS)与BORIS-N1 - 258结合,结合亲和力( Kd )为86.38 μM(图1D)。重组肽9(肽序列为RFDHWVWQSQPW)与BORIS-N1 - 258没有任何可测的亲和力(图1E)。肽2 ( DWSSWVYRDPQT )与BORIS-N1 - 258结合,弱结合亲和力( Kd )为314.5 μM (图1F)。为了验证相互作用,作者用生物素标记多肽9,并将其负载到SA传感器上,用一系列浓度的BORIS-N1 - 258测定Kd值,结果是5.30 nM。
BORIS-N1 - 258抗原用于选择从该菌中纯化出的肽9。为证实肽9与人细胞中BORIS-N1 - 258的相互作用,从不表达BORIS的HEK293细胞中表达并纯化了BORIS-N1 - 258。与肽9对细菌表达的BORIS抗原( Kd = 5.30nM )的亲和力相比,肽9与人细胞源性BORIS-N1 - 258结合,Kd值为6.37 nM(图 1G和 1H)。
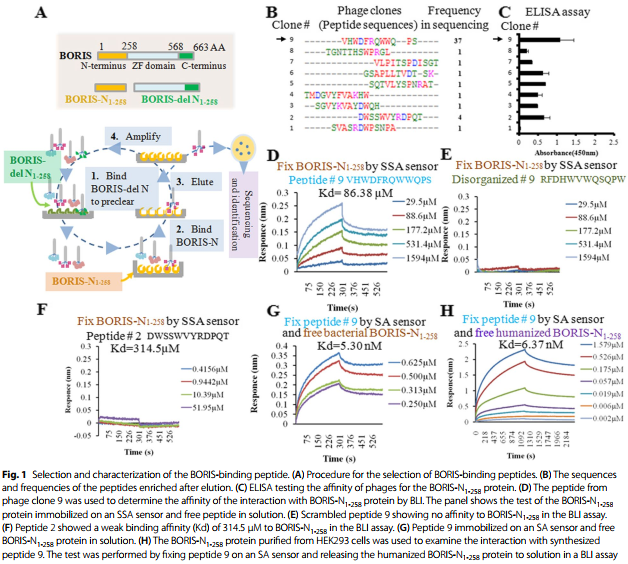
图1 BORIS - binding肽的特征和选择
2. 所选择的BTApep-TAT肽与BORIS在细胞中特异性结合
合成了多肽9与HIV - 1 TAT融合肽(肽序列为 GGRKKRRQRRRG),并用生物素标记。融合HIV- TAT肽赋予了穿透细胞膜的能力。本研究将融合HIV-1 TAT的生物素化肽9定为BTApep- TAT-biotin。合成了有生物素标记但缺乏穿透能力的肽( BTApep-biotin )来评估穿透细胞膜的效率。用含有12个连续组氨酸与HIV-1 TAT肽融合的生物素化肽作为阴性对照肽( His – TAT – biotin )。免疫共沉淀法检测BTApep - TAT - biotin与BORIS的相互作用。BTApep- TAT-biotin与全长BORIS和BORIS-N1 - 258结合,而不能与BORIS-delN1 - 258结合(图2A)。用siRNA ( siBORIS _ 1 )敲除H1299细胞中的BORIS后,商业BORIS抗体与BTApep- TAT-biotin肽的结合能力降低,表明BTApep-TAT能够特异性结合细胞中的BORIS(图2B)。

图2 所选择的BTApep - TAT肽与BORIS在细胞中特异性结合,诱导癌细胞DNA损伤和凋亡
3. BTApep - TAT诱导癌细胞DNA损伤和凋亡
采用BTApep - TAT梯度稀释法处理H1299肺癌细胞,进行MTT检测和细胞计数。结果发现,BTApep- TAT在25 ~ 100 μM浓度下处理3天后,H1299细胞的增殖受到抑制(图2C)。此外,BTApep- TAT抑制H1299细胞的增殖,而非正常HEK293细胞,BORIS不表达。BTApep不穿透细胞膜,不抑制细胞增殖(图2D)。
为探讨BTApep - TAT对BORIS的靶向性,采用高通量RNA测序方法比较了BTApep - TAT处理H1299细胞和2个siRNA敲除BORIS后的转录组细胞。BTApep - TAT处理和BORIS敲除样本间基因表达谱的热图比较如图2E所示。KEGG通路分析显示了与1004个共同基因相关的顶端通路(图2F)。在H1299细胞中,caspase-3/7实验结果表明BTApep-TAT诱导细胞凋亡,TUNEL实验结果表明BTApep-TAT引起DNA损伤(图2G和2H)。因此,BTApep- TAT处理可能会减弱BORIS对癌细胞基因组稳定性的保护作用。
4. BTApep - TAT抑制癌细胞中BORIS调控的DNA损伤修复
环境和内部应力,如诱变性化学物质、电离辐射( IR )、活性氧( ROS )和mis-replication 压力,诱发DNA损伤,包括DNA单链断裂( SSBs )和双链断裂( DSBs )。DNA修复受损会积累DNA病变,导致基因组不稳定。DNA修复异常促进癌症进展。即:BTApep - TAT诱导DNA损伤,减弱了BORIS对癌细胞基因组的保护作用。本研究将转染全长BORIS或BORIS-N1 - 258的HeLa细胞的粗核提取物与含有DNA损伤的模板和对照模板的混合物进行无细胞实验。BORIS-N1 - 258与BORIS相同程度地增强了SSBR活性(图3A),表明BORIS的AA1 - 258N末端负责BORIS的SSBR活性。接下来,在表达BORIS的HeLa细胞的粗核提取物中加入BTApep - TAT或His - TAT (阴性对照肽)。BTApep- TAT显著抑制SSBR和BER,而不抑制His- TAT(图 3B和 3C)。这些结果也表明BTApep- TAT而不是His- TAT诱导癌细胞DNA损伤和凋亡(图 2G和 2H)。
作者采用基于XhoⅠ线性化质粒的体外DNA末端连接实验评估了BORIS和BTApep- TAT在NHEJ中的功能。相关数据表明,BORIS促进了高效的末端加入。BTApep- TAT,而非His- TAT,处理抑制了约30 % BORIS诱导的末端连接(图3D)。BORIS-RFP转染细胞与未转染细胞进行DNA损伤修复的细胞百分比的比较如图3E所示。流式细胞仪对细胞进行计数和分析。总的来说,这些实验结果表明BORIS促进了癌细胞中SSB和DSB的修复,BTApep- TAT抑制了由BORIS控制的DNA损伤修复。
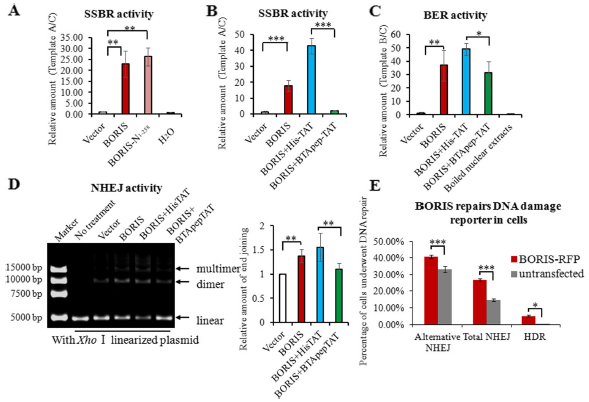
图3 BTApep - TAT抑制癌细胞中BORIS调控的DNA损伤修复
5. BTApep - TAT抑制BORIS的ADP核糖化来应对DNA损伤
HeLa和H1299细胞转染C端Myc-tagged BORIS。用抗ADP-核糖抗体免疫沉淀(同时识别多聚和单聚ADP-核糖基化)表明BORIS-myc被ADP-核糖修饰(图4A)。我们利用纯化的BORIS - N1 - 258蛋白和生物素标记的PAR聚合物进行了体外ADP核糖基化实验,证实BORIS的ADP核糖基化作用。生物素标记的PAR与BORIS - N1 - 258在体外呈浓度依赖性结合(图4B)。然后用抗ADP核糖抗体对转染BORIS-N1 - 258 - myc的H1299细胞进行免疫沉淀,证实了BORIS-N1 - 258 - myc的修饰作用。BORIS-N1 - 258 - myc的ADP核糖基化水平与全长BORIS ( BORIS-myc )相当(图4C)。这些结果证明了BORIS在氨基酸1 ~ 258中ADP核糖基化。
其次,研究了DNA损伤条件下BORIS的ADP核糖基化水平。合成双链DNA ( dsDNA )用于模拟DNA损伤细胞中的DNA断裂。将合成的dsDNA加入到BORIS表达细胞的裂解液中,检测其ADP核糖基化水平,发现dsDNA能促进BORIS在HeLa和H1299细胞中的ADP核糖基化(图4D)。此外,IR(照射)轻微诱导BORIS的ADP核糖基化(图4E),H2O2诱导BORIS的ADP核糖基化相当大(图4F)。X射线照射产生的SSB比DSB多25倍。这些结果提示dsDNA或ssDNA诱导BORIS发生ADP-核糖基化的幅度存在差异。虽然ssDNA和dsDNA均促进了BORIS的ADP核糖基化,但dsDNA的诱导作用强于ssDNA (图4G)。
将纯化的BORIS-N1 - 258蛋白分别与25 μM和50 μM的BTApep- TAT或His- TAT预孵育后进行体外ADP核糖基化实验,发现BTApep- TAT能有效抑制BORIS-N1 - 258的ADP核糖基化(图4H)。转染H1299细胞的BORIS-myc的修饰也被25 μM BTApep- TAT处理而不被His- TAT处理所抑制(图4I)。这些结果表明,BTApep- TAT无论在体外还是在体内都明显抑制了BORIS的ADP核糖基化。
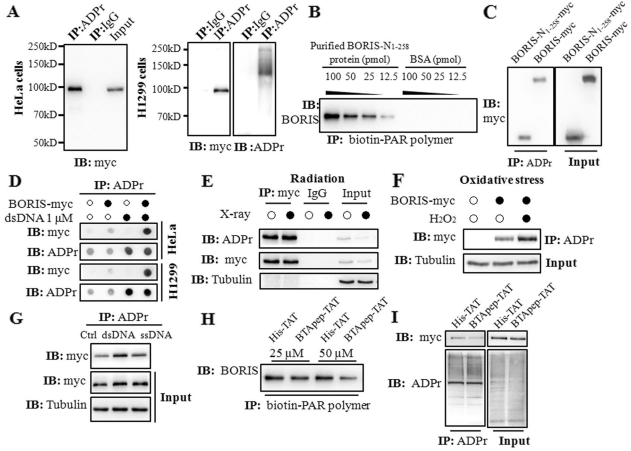
图4 BTApep - TAT抑制BORIS的ADP核糖化来应对DNA损伤
6. BTApep - TAT抑制BORIS 第198 - 228位的ADP核糖基化,阻断了BORIS与Ku70的相互作用
假设BORIS-N1 - 258的功能需要ADP核糖基化,为了确定BORIS中ADP核糖基化的位点,作者制作了几个截断突变体。缺失AA2 - 227产生了与AA228 - 663相对应的截短BORIS蛋白,与缺失AA2 - 197产生的AA 198 - 663 BORIS相比,该截短产物ADP核糖基化明显减少。这一观察表明,改性的主要位点位于残留物198 – 228(图 5A和 B)之间。已知谷氨酸近端序列为ADP核糖基化。利用ADPredict软件进行区域和站点的预测BORIS的N个区域,谷氨酸残基198、204、214、225和226是推测的ADP核糖基化位点,在小鼠、大鼠和人类中进化保守(图5B)。因此,将该区域内的5个谷氨酸( E )残基全部替换为丙氨酸残基( A ),产生了BORIS的五元组突变体( 5EA )(图5B)。5EA突变降低了BORIS的ADP核糖基化(图5C)。虽然5EA突变并未完全取消BORIS的ADP核糖基化,但观察到明显下降。BORISN1 - 258内另外两个潜在的ADP-核糖基化区( AA 29 - 33和 AA 165 - 185 )也通过丙氨酸残基替换谷氨酸残基而发生突变;然而,这些突变并没有减弱ADP核糖基化(图5D)。如图5C和D所示,5EA突变体降低了表观分子量,而E29 - 33A或E165 - 185突变体降低不明显。
BORIS-N198 - 228肽的多聚ADP核糖基化与其丰度成比例累积(图5E)。BORIS-5EA在体内照射或体外DNA连接实验中对DNA修复无活性(图5F-G)。当细胞与BTApep- TAT孵育后,BORIS与Ku70的相互作用被阻断(图5H)。BORIS - 5EA仅与Ku70弱相互作用,表明BORIS与Ku70结合需要ADP核糖基化(图5I)。综上所述,BORIS的ADP核糖基化与Ku70和DNA修复有关。BTApep- TAT通过干扰BORIS的ADP核糖基化及随后与Ku70的相互作用,减弱了BORIS在DNA损伤修复中的功能。
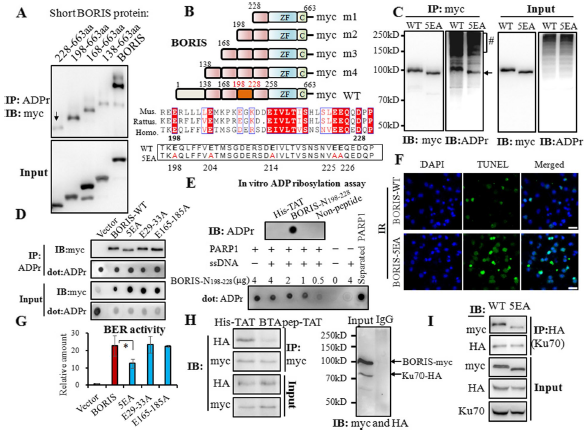
图5 BTApep - TAT抑制BORIS 198 - 228位点的ADP核糖基化,阻断了BORIS与Ku70的相互作用
为了验证BTApep - TAT在体内的功能,本研究利用体外对BTApep - TAT敏感的H1299细胞,建立NOD / SCID / γ c null ( NSG )小鼠移植瘤模型。12只小鼠前肢皮下注射H1299细胞(1×106/只)。1周后,将小鼠分为2组(每组6只),隔天腹腔注射16mg / kg BTApep- TAT或His- TAT,连续3周。隔日对肿瘤进行评估。安乐死和肿瘤切除后,于实验结束时测量肿瘤重量(图6A)。与His- TAT治疗相比,BTApep- TAT抑制肿瘤生长(图6B)。此外,BTApep - TAT处理未引起肝、肾毒性(图6C)。肿瘤切片检查采用TUNEL法。用BTApep- TAT治疗3周,而不用His- TAT,诱导皮下肿瘤细胞DNA损伤(图6D)。
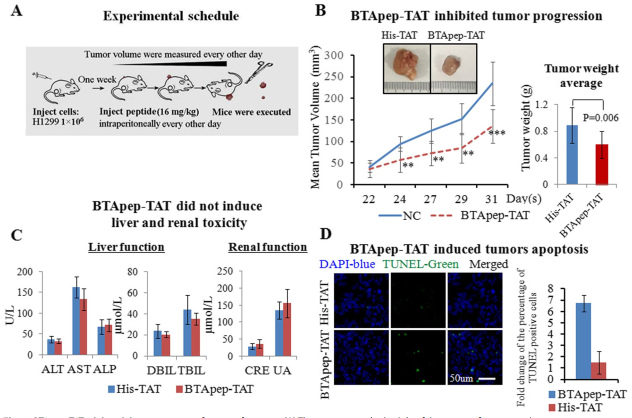
图6 BTApep - TAT可抑制皮下肿瘤的进展
结论
本研究表明BTApep- TAT肽可以靶向BORIS。BTApep - TAT与BORIS相互作用,抑制BORIS相关转录组,模拟BORIS敲减的效应。BTApep- TAT抑制癌细胞增殖,诱导癌细胞凋亡,抑制移植瘤模型中NSCLC进展,抑制BORIS的ADP核糖基化。BORIS在198 - 228残基上的ADP核糖基化促进了与Ku70的相互作用,并负责DNA修复。研究结果表明了BTApep- TAT肽用于癌症治疗的可行性,为今后深入剖析BORIS的调控机制和设计优化的BORIS抑制剂提供了依据。
参考文献
Zhang Y, Fang M, Li S, Xu H, Ren J, Tu L, Zuo B, Yao W, Liang G.(2022) BTApep-TAT peptide inhibits ADP-ribosylation of BORIS to induce DNA damage in cancer. Mol Cancer. 21(1):158. doi: 10.1186/s12943-022-01621-w.