






线粒体和铁死亡——GPD2协同线粒体GPX4抵御铁死亡
我们对细胞器抵御铁死亡的机制了解甚少,这阻碍了我们在疾病治疗中通过靶向铁死亡达到治疗目的。本研究通过代谢组分析发现GPX4抑制剂处理后导致细胞中的甘油(G3P)丢失。进一步研究大小补充G3P的癌细胞以G3P脱氢酶2 (GPD2)依赖的方式减弱由GPX4抑制剂诱导的铁死亡,GPD2缺失使得癌细胞对GPX4抑制引起的线粒体脂质过氧化和铁死亡敏感,并且GPX4和GPD2同时敲低能协同抑制铁死亡引起的肿瘤生长。这些结果表明GPD2参与线粒体抵御铁死亡。本研究于2022年6月发表在《Proc Natl Acad Sci USA》IF:12.779期刊上。
技术路线:

主要实验结果:
1、代谢组分析发现GPD2与铁死亡调控关联
在GXP4急性失活即用GPX4抑制剂处理后的2小时来挖掘铁死亡的抵御机制。代谢组分析发现在肿瘤细胞用GPX4抑制剂(RSL3,ML210,ML162)处理2小时后 C-Asp,一种嘧啶生物合成的中间产物,是最显著下调的,此外还发现G3P也在3种抑制剂处理的癌细胞中显著下降了(图1A-D)。而G3P补充显著抑制RSL3诱导的不同癌细胞系的铁死亡(图1E)。这表明G3P具有抵御GPX4失活引起的铁死亡的能力。
于是进一步探究G3P关联铁死亡调控的代谢酶,如图1F所示,线粒体内膜结合的GPD2将G3P转换回DAP,并将电子传递给线粒体中的电子传递链。Cancer Therapeutics Response Portal(CTRP)数据分析发现GPD2的表达而非GPD1,GPD1L,GK,PGP与细胞对GOX4抑制剂抗性相关(图1G-H)。因此,后续研究关注GPD2。
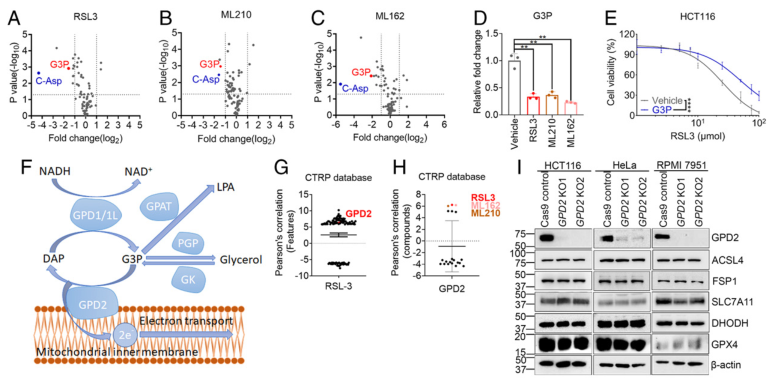
图1代谢组分析发现GPD2与铁死亡调控关联
为了探究GPD2在铁死亡调控中的作用,采用CRISPR-Cas9方法分别在宫颈癌HeLa细胞,皮肤癌RPMI7951细胞,和大肠癌HCT116中敲低GPD2的表达,结果发现GPD2敲低不影响基底细胞活力但使得这些癌细胞对GOX4抑制剂诱导的细胞死亡和脂质过氧化敏感(图1J-1S)。在HeLa和RPMI7951细胞中,细胞死亡被铁死亡抑制剂ferrostatin-1或铁螯合剂(DFO)完全抑制,证实这些GPX4抑制剂诱导了这些细胞系的铁死亡(图1K, L, N, O),然而HCT116细胞在用ferrostatin-1或DFO处理后仍有残留死亡(图1M, P),表明这些GPX4抑制剂主要触发铁死亡,但也可诱导HCT116细胞的其他非铁死亡的细胞死亡。此外,GPD2缺失似乎不影响GPX4、FSP1、dhdh、SLC7A11、ACSL4等其他铁死亡调控因子的表达(图1I)。最后,研究显示G3P抵御RSL3诱导细胞死亡的保护作用被GPD2敲除所废止(图1T-W)。综上所述,这些数据表明G3P通过GPD2和GPD2缺乏使细胞对GPX4抑制引发的铁死亡敏感保护细胞免受GPX4抑制引起的铁死亡。

图2 GPD2敲除促进GPX4失活诱导的铁死亡
GPD2是定位在线粒体内膜上的酶。分级分析证实了内源性GPD2在几种癌细胞系中的线粒体定位(图3A)。与此前的研究一直,MitoPerOx染色显示RSL3处理不导致强烈的线粒体脂质过氧化,重要的是,GPD2敲低显著增强RSL3处理下的线粒体脂质过氧化(图3B-E)。野生型GPD2在GPD2敲除细胞中的恢复,而不是其线粒体定位缺陷的突变体(42AA),挽救了GPD2缺陷对RSL3诱导的细胞死亡和线粒体脂质过氧化的影响(图3A,F-L)。此外,在GPD2野生型细胞中,使用自由基捕获抗氧化剂TEMPO或ferrostatin-1处理完全抑制了RSL3诱导的细胞死亡,但使用Mito-TEMPO不能提供任何保护作用(图3M-P)。相反,在GPD2-KO细胞中,Mito-TEMPO和ferrostatin-1及TEMPO的作用类似,可以完全抑制RSL3诱导的线粒体脂质过氧化(图3Q-S),并显著抑制细胞死亡(图3T-W)。综上所述,这些数据表明GPD2抑制线粒体脂质过氧化,而RSL3在GPD2缺陷细胞中诱导的铁死亡至少部分是由线粒体脂质过氧化引发的。

接下来探究GPD2抑制线粒体脂质过氧化和铁死亡的机制。分析发现GOD2敲除使细胞对GOX4抑制剂RSL3和ML210敏感(图2J-P),但是对另一种铁死亡抑制剂FIN56,其可耗竭CoQ和GPX4(图4A-D)。由于GPX4抑制剂和FIN56之间的主要区别在于它们对CoQ耗尽的影响不同,这些数据表明GPD2以CoQ依赖的方式抑制铁死亡(例如,当CoQ被FIN56耗尽时,GPD2敲除对铁死亡的作用就减弱了)。与此一致,作者发现在用4-氯苯甲酸(4CBA)处理阻断CoQ合成的条件下, GPD2敲除的铁死亡敏感效应被消除(图4E-F)。同样地,COQ2缺失消除了GPD2缺失对RSL3诱导的铁死亡的敏感作用(图4G-H)。这些结果与已知的CoQ抑制脂质过氧化和铁死亡作用相符。在线粒体内膜中,GPD2将G3P氧化为DAP,并将CoQ还原为CoQH2(图1F)。作者证实GPD2敲除导致HeLa 和HCT116e细胞中CoQ/CoQH2比例显著增加(图4I-J)。此外,补充MitoQH2(线粒体靶向CoQH2)完全抑制了RSL3处理的GPD敲除细胞的线粒体脂质过氧化(图4K-M)并废除了GPD2导致的铁死亡敏感性(图4N-P)。这些数据表明在线粒体中GPD2通过还原CoQ至CoQH2抑制脂质过氧化和铁死亡。
近来的研究表明,FSP1和DHODH分别通过在质膜和线粒体中产生CoQH2来抑制铁死亡。因此,作者进一步探究GPD2与DHODH或FSP1在调控铁死亡中的相互作用。过表达DHODH而非FSP1,可以显著挽救GOD2缺失细胞的铁死亡敏感表型(图4Q-R),此外,GPD2过表达可以部分挽救DHODH缺失细胞的铁死亡敏感表型(图4U-V)。GPD2 与线粒体中的 DHODH 并行运行,但不与质膜上的 FSP1 并行运行,以防止铁死亡。
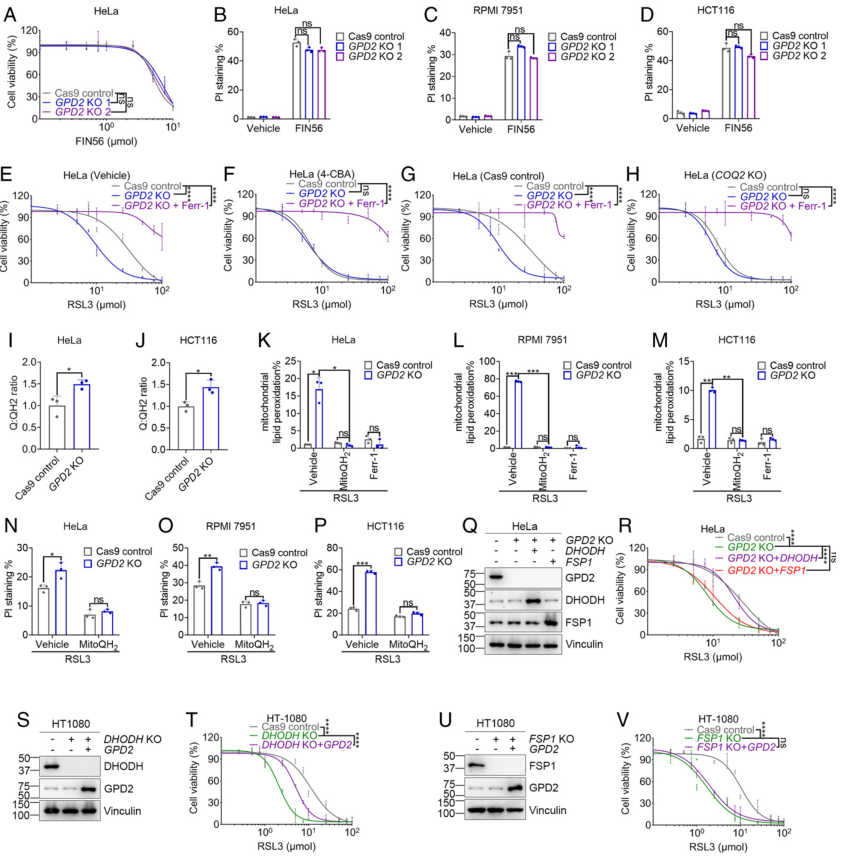
图4在线粒体中GPD2通过还原CoQ至CoQH2抑制铁死亡
5、在体外细胞模型中GPD2与线粒体GPX4协同作用抑制铁死亡
上述结果促使作者继续研究铁死亡调控中GPD2和GPX4互作。为此,利用CRISPR-Cas9方法构建了GPD2和GPX4单敲除(KO)和双敲除(DKO)的HCT116细胞(图5A)。研究显示GPX4-KO HCT116细胞在无ferrostatin-1培养基中长时间培养(14 ~ 15 d)后完全死亡(图5B)。GPD2缺失不影响GPX4野生型细胞的活力,但显著加速了GPX4-KO细胞的死亡,如在无ferrostatin-1培养基中培养7d后大部分DKO细胞死亡(图5B-D)。值得注意的是,ferrostatin-1能完全抑制GPX4遗传消融引起的HCT116细胞死亡,却不能改变RSL3处理引起的死亡,说明RSL3具有脱靶效应,导致HCT116细胞非铁死亡性细胞死亡(图2M vs 图5B-D)。一致地,DKO细胞的脂质过氧化水平明显高于GPX4-KO细胞(图5E)。在去除ferrostatin-1后,DKO细胞而非单KO细胞表现出强烈的线粒体脂质过氧化(图5F)。
有几种 GPX4 亚型具有不同的亚细胞定位,包括细胞溶质和线粒体 GPX4。 作者在GPX4-KO或DKO细胞中恢复细胞溶质或线粒体GPX4,分级分析证实,细胞溶质和线粒体GPX4正确定位于各自的隔室(图5G)。随后发现,过表达细胞溶质GOX4而非线粒体GPX4显著抑制了GPX4-KO细胞的脂质过氧化和铁死亡(图5H-I)。与此形成鲜明对比的是,细胞溶质GPX4未能抑制DKO细胞的铁死亡性细胞死亡(图4J),而线粒体GPX4的过表达显著推迟了DKO细胞的死亡,并且这种DKO细胞的死亡动力学与线粒体GPX4恢复似乎与GPX4-KO 细胞的恢复相似(图5B vs图5J)。ferrostatin-1后的第5天(在DKO细胞经历铁死亡之前),线粒体GPX4的恢复显著抑制了DKO细胞中总脂质过氧化或线粒体脂质过氧化的诱导,但胞质GPX4的恢复无抑制作用(图5K-L)。总之,这些数据表明GPD2与线粒体GPX4平行作用,但不与胞质GPX4作用,以抑制线粒体脂质过氧化和铁死亡。
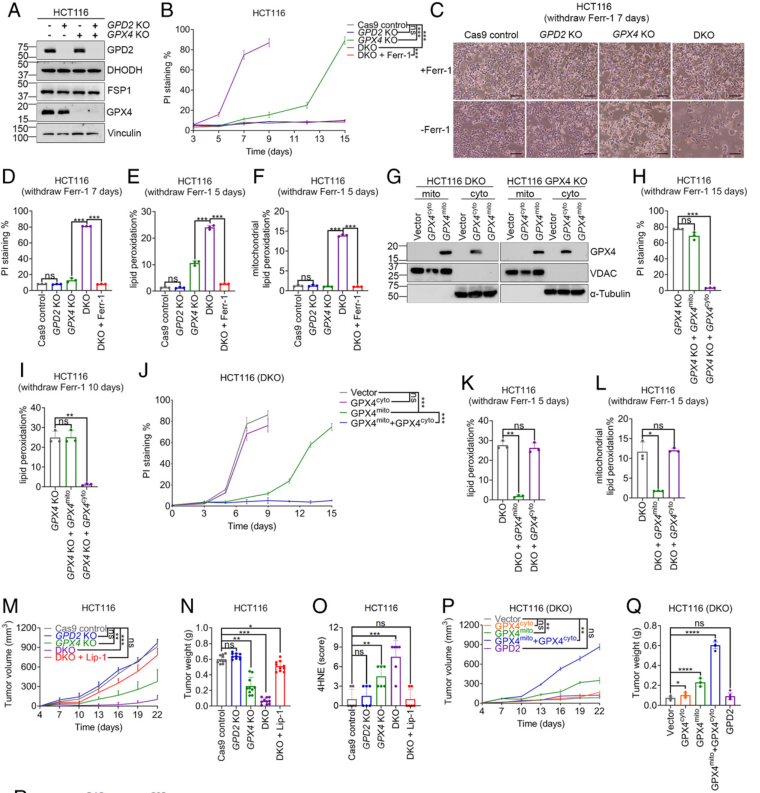
图5 GPD2与线粒体GPX4协同作用抑制细胞铁死亡
6、GPD2与线粒体GPX4协同抑制肿瘤铁死亡
最后作者在体内探究GPD2和/或GPX4失活是否通过诱导铁死亡影响肿瘤生长。由于可获得的GPD抑制剂(iGP-1)在本系统模型中无法发挥作用,并且没有适用于动物模型的GPX4抑制剂,所以作者采用了基因学的方法解决该问题。注射对照、GPX4-KO、GPD2-KO、或DKO的HCT116细胞至裸鼠中,然后监测肿瘤生长。结果发现GPX4-KO而非GPD2-KO显著抑制肿瘤生长,然而DKO组表现出更显著的肿瘤生长抑制,重要的是铁死亡抑制剂liproxstatin-1处理几乎完全恢复了DKO组肿瘤生长(图6M-N)。进一步的分析显示,尽管cleaved caspase-3和Ki67染色在不同基因型或处理之间没有显著差异,但脂质过氧化标记物4-HNE的表达,与对照组和GPD2-KO肿瘤相比,在DKO组水平更高,使用liproxstatin-1治疗后,4-HNE染色水平完全恢复到对照组的水平(图6O)。在DKO背景下,线粒体GPX4的恢复部分回复了肿瘤生长,同时恢复胞质和线粒体GPX4完全使肿瘤生长回复到与对照肿瘤相似的水平,而恢复细胞质GPX4或GPD2对肿瘤生长没有影响(图6P-Q)。这些体内结果支持了前面的体外研究,即GPD2与线粒体GPX4协同抑制肿瘤铁死亡。
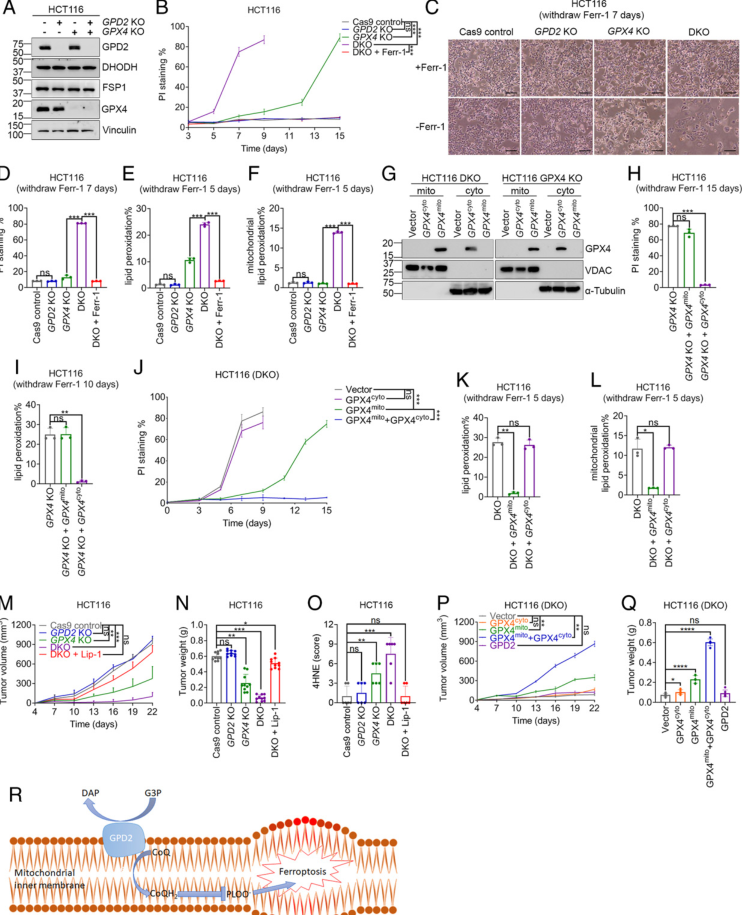
综上所述,本研究证实GPD2通过在线粒体内膜生成CoQH2来抑制线粒体脂质过氧化,从而抵御细胞铁死亡(图6R)。
参考文献:
Wu Shiqi., Mao Chao., Kondiparthi Lavanya., Poyurovsky Masha V., Olszewski Kellen., Gan Boyi.(2022). A ferroptosis defense mechanism mediated by glycerol-3-phosphate dehydrogenase 2 in mitochondria. Proc Natl Acad Sci U S A, 119(26), e2121987119. doi:10.1073/pnas.2121987119