






TBX2通过与CoREST复合物相互作用,作为肿瘤抑制基因的有效转录沉默者,以维持乳腺癌的增殖
TBX2基因位于17q23,常在乳腺肿瘤亚群中过表达。TBX2是一种抗衰老基因,通过抑制肿瘤抑制基因(TSGs),如NDRG1和CST6,促进细胞生长和存活。作者前期研究发现TBX2与PRC2复合体协同抑制多个TSGs,PRC2抑制恢复NDRG1表达从而抑制细胞增殖。在这里,作者现在确定CoREST蛋白LSD1和ZNF217是TBX2的新型互作蛋白。CoREST的遗传或药理学靶向模拟TBX2缺失,诱导NDRG1表达,并在体外和体内消除乳腺癌生长。此外,作者发现TBX2/CoREST靶向NDRG1是通过Sp1招募TBX2到NDRG1启动子上,从而导致NDRG1上调和癌细胞增殖减少来实现的。通过ChIP-seq,作者发现30 %的TBX2结合启动子与ZNF217共享,并鉴定出TBX2/CoREST抑制的新靶标;在这些靶点中,lncRNA LINC00111作为细胞增殖的负调控因子。总的来说,这些数据表明抑制CoREST蛋白对TBX2成瘾的乳腺肿瘤是一种有前途的治疗干预。本研究于2022年6月发表于《Nucleic Acids Res》,IF:19.160。
技术路线:

主要研究结果:
1、LSD1与TBX2相互作用,是乳腺癌细胞存活所必需的
为确定TBX2是否可以形成除PRC2-like复合物以外的其他抑制复合物,作者开展下面的研究。首先,在TBX2依赖的MCF7和T47D乳腺癌模型中,使用56个已知表观遗传修饰的核糖核酸内切酶siRNA(esiRNA)库进行活性筛选;任何导致细胞活力降低的“hits”都可以代表TBX2潜在的功能互作蛋白(图1A)。取4个“hits”(Hdac7、Sirtuin-3(SIRT3)、LSD1和Jumonji Domain-Containing 2B(JMJD2B)/KDM4B),对其在两种细胞系的活力进行下游验证。用两个独立的siRNA敲低每一个候选基因,48 h后的细胞活力实验发现,SIRT3和LSD1对MCF7细胞活力明显增强,但在MCF10A正常乳腺细胞中没有(图1B-C)。然而,在这些短期敲低中,LSD1敲低对TBX2靶标CST6的mRNA上调表现出最显著和可重复的效应,与CST6通常抑制的酶Legumain活性降低相一致(图1D-E)。这表明LSD1可能是TBX2可能的互作伙伴。免疫共沉淀实验证实,在MCF7和T47D模型中,TBX2和LSD1之间存在物理相互作用(图1F)。这些数据表明TBX2可能与多种表观遗传调控因子相互作用,抑制LSD1可作为靶向TBX2依赖性乳腺癌的可行策略。
作者下面确定TBX2-LSD1相互作用是否存在转录复合体中转录因子与其他染色质调控蛋白的相互作用影响蛋白的稳定性。首先,靶向TBX2的siRNA处理MCF7细胞72h后,细胞增殖能力降低,同时伴随着靶基因NDRG1和CST6的上调;然而,Western blot分析显示这独立于对LSD1蛋白水平的影响(图2A)。同样地,用两个独立的siRNA敲低LSD1,TBX2的敲低效果类似,但TBX2的RNA和蛋白没有相应的减少(图2C)。克隆形成实验也证实,LSD1和TBX2敲低对长期生存的影响相当,克隆数显著减少(图2B-D)。这些现象在第二个细胞系(T47D)中也是一致的,敲低TBX2或LSD1导致类似的细胞增殖减少和NDRG1/CST6上调,而在蛋白水平上不影响彼此的稳定性(图2E)。结晶紫生长实验再次证实TBX2和LSD1是T47D细胞克隆存活所必需的(图2F)。综合这些数据表明,虽然TBX2和LSD1对蛋白稳定性没有相互要求,但两者的相互作用可能对抑制TBX2靶基因和维持乳腺癌细胞生长具有重要意义。

图1 LSD1与TBX2相互作用。
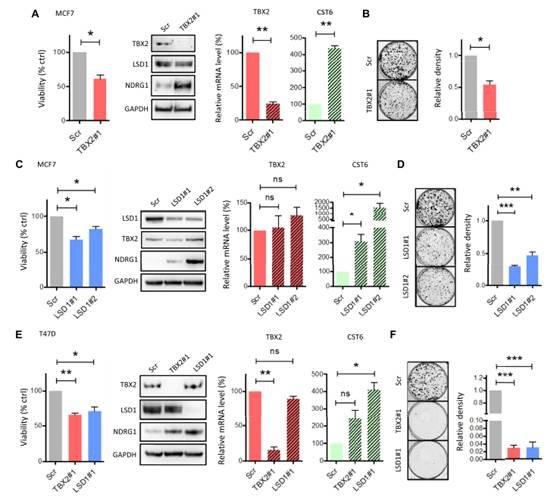
图2 LSD1敲低TBX2表型缺失。
2、变构LSD1抑制剂SP-2509在体内抑制TBX2靶点并抑制雌激素依赖的乳腺肿瘤生长
在确定LSD1是TBX2的一个新的细胞增殖所需的相互作用因子后,作者通过检测LSD1抑制剂SP-2509、RN-1和GSK-LSD1来研究这种依赖性是否可以被药理学靶向。首先,MCF7和T47D细胞暴露于浓度递增的LSD1抑制剂(图3A)。在测试的3个抑制剂中,只有SP-2509显示出降低细胞增殖的效果,72 h时对MCF7的IC50为250 nM,对T47D的IC50为1 M。两株细胞均对RN-1和GSK-LSD1的作用高度耐药,在最大20 M剂量下无法达到IC50;事实上,GSKLSD1确实促进MCF7(可能是由于脱靶效应)的生长。Western blot结果显示,IC50剂量的SP-2509处理后,NDRG1的表达显著上调,而20 M RN-1或20 M GSK-LSD1则无此作用。与作者之前观察到的LSD1敲低后TBX2的表达情况类似,SP-2509干扰LSD1的活性后TBX2的蛋白水平没有受到影响。在MCF7细胞中,NDRG1的上调与H3K4二甲基化(H3K4me2)的整体增加相关,而在T47D细胞中,NDRG1的上调与该组蛋白标记不相关,表明H3K4me2的整体变化是SP-2509处理的间接效应(图3B)。无论H3K4me2水平如何,用SP-2509 IC50处理两种细胞系均导致靶基因NDRG1和CST6的转录显著上调,而RN-1或GSK-LSD1在20M时几乎没有影响(图3C-D)。相应地,SP-2509处理导致CST6靶点Legumain的活性显著降低(图3E)。接下来,作者利用Tet-off诱导模型探讨SP-2509是否可以发挥TBX2依赖的表型;该基因表达一个显性负调控TBX2蛋白(DN-TBX2),由包含完整T结构域的1-301个氨基酸组成,但缺少抑制结构域所在的302-701个氨基酸。有趣的是,SP-2509的抗增殖作用在诱导表达截短型TBX2的MCF7细胞中更加明显(图3F),同时伴随着靶标NDRG1和CST6的上调,显著高于单独处理组(图3F)。值得注意的是,DN-TBX2对LSD1蛋白表达的影响可以忽略不计,这表明这种对SP-2509的敏感性增强不是由于药物靶标的下调。为确定SP-2509是否能在体内抑制乳腺肿瘤的生长,用MCF7细胞移植雌激素补充的小鼠,用40 mg/kg的SP-2509或溶剂处理4周(图3G)。终点免疫组织化学染色发现,SP-2509处理的肿瘤组织中增殖标志物Ki-67的表达约为溶剂对照组的一半(图3H)。总的来说,这些体外和体内的数据表明,SP-2509特异性靶向LSD1的H3变构位点,可以导致TSGs的有效上调,并可能是用于开发乳腺癌中的TBX2依赖性的一个可行的治疗方案。
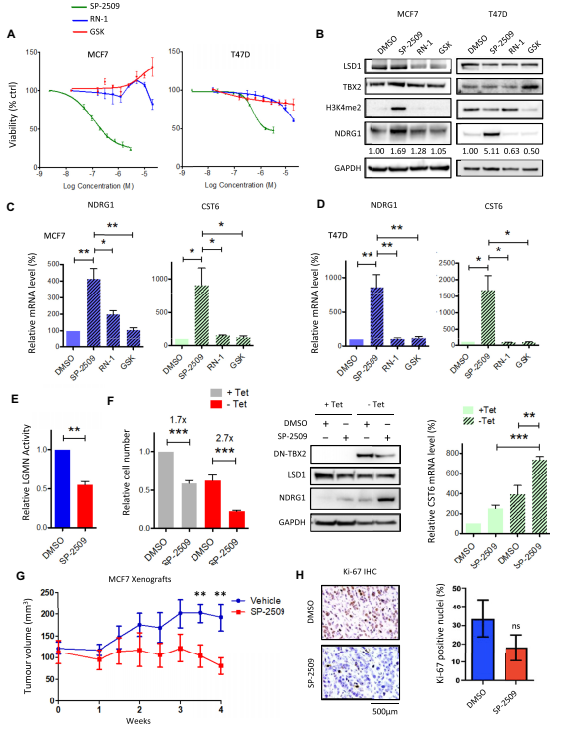
图3 一种变构LSD1抑制剂增强tbx2抑制基因的表达,并在体内阻止乳腺肿瘤的生长。
3、TBX2与CoREST复合体的组件相互作用
对TBX2敲低后上调的转录本进行ChEA富集分析,发现ZNF217是第二显著的TF结合位点(图4A)。因此,作者推测TBX2可能与ZNF217相互作用,除LSD1外,还可能与CoREST复合体的其他成员相互作用。因此,从MCF7和BT474细胞提取物中免疫沉淀ZNF217,证实了除CoREST组分LSD1和HDAC1外,还与TBX2存在物理相互作用(图4B)。这表现在TBX2的下拉互作中,表现为与ZNF217、LSD1和HDAC1的正向互作(图4C),重要的是,这些数据再现之前的发现,TBX2可以与HDAC1相互作用,这被证明依赖于TBX2的C端。
通过两个独立的ZNF217 siRNA序列处理TBX2依赖的细胞,评估ZNF217对TBX2依赖的细胞存活的重要性,这导致目标NDRG1和CST6的生长显著降低和转录上调(图4D)。与LSD1一样,ZNF217敲低后NDRG1的上调与TBX2蛋白表达的变化无关,反之亦然(图4E)。在BT474和T47D乳腺细胞系中进一步证实了这种依赖性,其中ZNF217的敲低导致NDRG1显著上调并显著降低克隆形成率(图4F)。
为了进一步评估CoREST依赖性,通过增加抑制剂MS-275(恩替诺特)的剂量处理表达TBX2的细胞72 h,证实其对I类HDACs的敏感性,其在MCF7和T47D细胞中的IC50分别为1.5 M和0.5 M。由于多个CoREST相关蛋白的敲除可以解除对TBX2靶点的抑制,因此提出同时抑制多个复合物成员应该导致明显的表型。因此,作者发现低剂量的SP-2509和恩替诺特(IC50以下)对NDRG1/CST6的生长抑制和转录上调作用明显大于单药处理(图4G)。这些结果表明,TBX2与CoREST蛋白LSD1、ZNF217和HDAC1之间的相互作用和协调活性可能是抑制TSGs促进乳腺肿瘤存活所必需的。

图4 TBX2与CoREST抑制复合物的组分相互作用。
4、TBX2和ZNF217显示出截然不同和重叠的全局DNA结合谱
基于作者的证据,TBX2与CoREST因子共同存在于抑制复合体中,作者进一步将作者的内部TBX2数据与公开的MCF7进行了比较该复合物中关键蛋白的ChIP-seq。由于缺乏高质量的LSD1公共数据以及作者无法通过甲醛方案实现LSD1富集,作者利用ZNF217 ENCODE ChIP-seq数据来代表潜在的CoREST靶向区域。作者发现30 %的TBX2结合位点与ZNF217峰中心重叠,这些峰的平均信号富集度在两种蛋白中高度相似(图5A)。与ZNF217重叠的TBX2峰主要见于启动子区域(图5B),激酶富集分析显示其富集于PDGFRA下游靶基因(图5C)。有趣的是,这些TBX2/ZNF217重叠峰对之前发现的CTCF和NF-Y基序缺乏富集,而对Fos/Jun(AP-1)、THAP11、GABPA和Sp1共有序列显示显著富集(图5D)。相反,ZNF217信号缺失的其余TBX2位点富集在Sp1、CTCF、NF-Y和CREM基序,而AP-1、THAP11和GABPA基序没有富集;综上表明ZNF217影响TBX2在AP-1、THAP11和GABPA位点的定位,而ZNF217本身不经常发生在TBX2结合的NF-Y、CTCF和CREM位点。相应地,对TBX2信号缺失的ZNF217结合位点分析发现,THAP11、AP-1、GABPA和CTCF基序显著出现,而NF-Y和CREM序列没有富集(图5D)。作者进一步检测TBX2和ZNF217的结合位点,其中包括CoREST复合物关键组分RCOR1的ENCODE公共ChIP-seq数据。三个蛋白均富集的靶基因可能代表TBX2-CoREST抑制基因。虽然部分TBX2/ZNF217共享区域显示重叠的RCOR1信号,但在ZNF217结合(图5E)的剩余TBX2位点中没有观察到中心RCOR1富集。这些数据突出表明ZNF217对于RCOR1定位到TBX2靶区域是必不可少的,并且发现了一组TBX2可能与CoREST蛋白相互作用以实现转录抑制的目标启动子。ZNF217对TBX2-CoREST抑制的重要性也可能解释为什么SP-2509破坏复合物中ZNF217的相互作用可以有效地去抑制TSGs和抑制乳腺癌细胞增殖。

图5 ZNF217与乳腺癌细胞中三分之一的TBX2结合位点重叠。
5、Sp1对于TBX2介导的NDRG1抑制至关重要
鉴于MCF7 ChIP-seq中TBX2结合位点最富集于Sp1基序,作者推测Sp1可能与TBX2介导的基因抑制有关。免疫共沉淀实验证实TBX2与Sp1存在相互作用(图6A)。接下来,作者检测NDRG1位点对Sp1识别位点的影响(图6B)。与其他在电子数据库中鉴定的TBX2/CoREST靶点相比,NDRG1位点的TBX2、ZNF217和RCOR1结合位点的排列遵循非典型模式;而在启动子TSS处存在明显的TBX2和RCOR1峰,该区域缺少一个较强的ZNF217峰。相反,ZNF217结合被发现富集在NDRG1位点3 '端内含子区域,与RCOR1共定位。重要的是,位于内含子10的区域被归类为“基因内增强子”,在GeneHancer双精英数据库中被编目以靶向NDRG1 TSS(图6B)。内含子10作为NDRG1的内部增强子的证据被公开的MCF7数据进一步证实,表明该区域在3D实验中与启动子存在物理相互作用;启动子和内含子也被富集到组蛋白修饰H3K27Ac和组蛋白H3第4位-甲基化,活性增强子的标志。因此,NDRG1启动子上缺乏强的ZNF217信号可以通过染色质环化来解释,从而影响蛋白质从一个复合物固定到两个明显不同的DNA区域。有趣的是,主上皮因子GRHL2(CCNGTTNNNCNAG)的基序在NDRG1启动子和第10内含子的ChIP峰中心都存在(图6B),而完全保守Sp1序列(GGGGCGGGGC)特异于TBX2峰的中心启动子处。鉴于之前的研究发现GRHL2和Sp1分别是参与ZNF217和TBX2在MCF7细胞中全局结合的关键基序,这些观察结果是拟合的(图5D)。有趣的是,TBX2与GRHL2存在物理互作(图6C)。然而,在TBX2依赖的细胞系中,单独敲低Sp1被认为足以使NDRG1上调(图6D),而该蛋白的丢失显著降低了长期生存(图6E)。匹配mRNA的分析也证实Sp1缺失后NDRG1的上调发生在转录水平(图6F);因此,这些数据表明Sp1可能对NDRG1的抑制和维持细胞增殖有重要作用。对作者的TBX2 ChIP-seq数据进行分析发现,Sp1基序主要位于TBX2峰顶100bp内,表明这两个因子之间存在协同或竞争的DNA结合(图6G)。在NDRG1位点,ChIP PCR显示Sp1敲低后TBX2与启动子/ TSS区域的结合明显减弱(图6H)。作者观察到siRNA敲低NDRG1部分挽救TBX2和ZNF217敲低的抗增殖作用(图6I-J)。这些结果表明TBX2-CoREST复合物对NDRG1的转录抑制是通过Sp1招募TBX2到NDRG1启动子上实现的。虽然NDRG1在TBX2/CoREST缺失后发挥抑癌作用,但其他一些靶基因(潜在的新型TSGs)可能参与了这一表型,因此需要进一步研究。
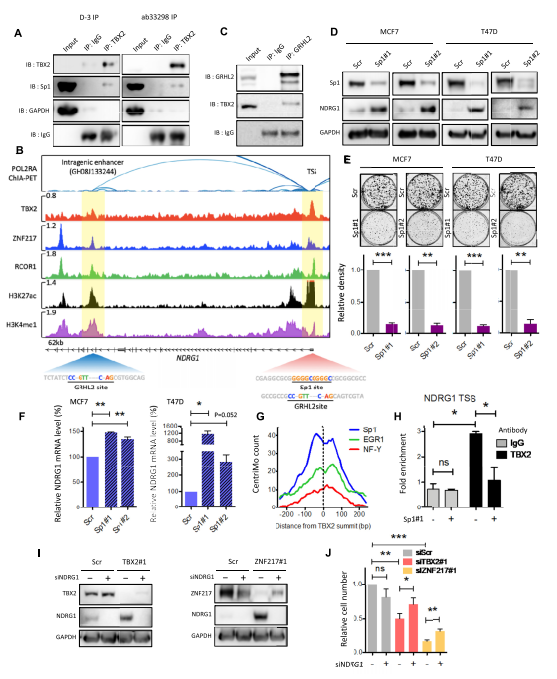
图6 Sp1通过招募TBX2到NDRG1启动子来抑制TSG NDRG1。
6、LINC00111被TBX2-CoREST抑制,在乳腺癌细胞中表现出肿瘤抑制活性
鉴于ZNF217在MCF7 ChIP-seq中占据了大量的TBX2结合位点,作者确定了这些区域中的哪些区域代表了TBX2-CoREST抑制基因,这些基因可能具有肿瘤抑制功能。在包含RCOR1的TBX2/ZNF217结合区域的子集中,作者筛选出7个启动子,它们的转录本因TBX2或ZNF217功能的丧失而持续上调;其中6个基因为蛋白编码(CELSR2、CORO2A、CTNND2、GOLT1A、KLHL20、PTK6),1个基因为长链非编码RNA产物(LINC00111)(图7A)。这些基因在TF结合位点内都至少含有一个之前发现的显著性基序(图5D)。LINC00111启动子与其他区域不同,TBX2峰含有CREM基序,在TBX2结合位点整体中较少出现(图7A)。作者发现这7个基因都受到TBX2-CoREST的转录抑制,用TBX2 siRNA或ZNF217/LSD1抑制剂SP-2509处理后,相关的RNA显著上调。LINC00111被发现是上调最强烈的转录本,其诱导倍数与NDRG1相当(图7B-C)。
作者为确定TBX2-CoREST抑制靶点是否在细胞生长和存活中发挥作用,在缺乏和存在LINC00111 siRNA的情况下敲低TBX2,比较对细胞生长的影响(图7D)。结果发现,LINC00111 siRNA处理显著挽救TBX2 siRNA以及HP1和KAP1敲低的抗增殖作用(图7D)。有趣的是,单独使用LINC00111 siRNA处理导致TBX2蛋白显著上调,这自然与TBX2 siRNA的作用相反(图7D,1);这暗示LINC00111本身可能作为TBX2的阻遏物。此外,LINC00111 siRNA基底降低了促衰老因子细胞周期蛋白激酶抑制因子P21Waf1/Cip1的蛋白水平,同时阻止了TBX2 siRNA对细胞周期蛋白激酶抑制因子P21Waf1/Cip1的上调作用(图7D,5、6)。LINC00111 siRNA不影响HP1 /KAP1的蛋白表达(图7D,2、3);然而,通过LINC00111 siRNA处理(图7D,5、6),HP1/KAP1敲低对细胞周期蛋白激酶抑制因子P21Waf1/Cip1的上调作用大大降低。相反,LINC00111 siRNA不能挽救EZH2敲低的抗增殖作用,也不能导致细胞周期蛋白激酶抑制因子P21Waf1/Cip1的上调(图7D,4、5、6)。这些数据表明LINC00111在与细胞周期蛋白激酶抑制因子P21Waf1/Cip1诱导相关的生长停滞中发挥重要作用。
为进一步证实LINC00111在生长阻滞中的作用,在MCF7和MDA-MB-361细胞中重复实验,其中TBX2在LINC00111 siRNA存在和不存在的情况下被敲低。RT-qPCR证实TBX2缺失后LINC00111表达上调,LINC00111 siRNA干扰后LINC00111表达下调。如前所述,LINC00111敲低引起TBX2本身的上调(图7E-F)。在这两种细胞系中,LINC00111敲低阻止了TBX2 siRNA对细胞周期蛋白激酶抑制因子P21Waf1/Cip1的上调,并部分挽救了增殖标志物CDK1的丢失(图7F)。在长期实验中,LINC00111敲低也显著降低了TBX2 siRNA对克隆形成的不利影响(图7G)。总的来说,这些结果表明LINC00111通过调节TBX2和细胞周期蛋白激酶抑制因子P21Waf1/Cip1的表达而具有肿瘤抑制特性,因此当被TBX2-CoREST复合物靶向抑制时,对癌细胞存活具有重要的影响。

图7 LINC00111是TBX2-CoREST的转录抑制靶点,具有抑瘤活性。
结论
总之,作者鉴定了一个由TBX2抑癌基因形成的新的转录复合物。TBX2对其他转录因子的重新配置以及与多个抑制复合物的界面的适应性使其成为一个非常强大的致癌基因。
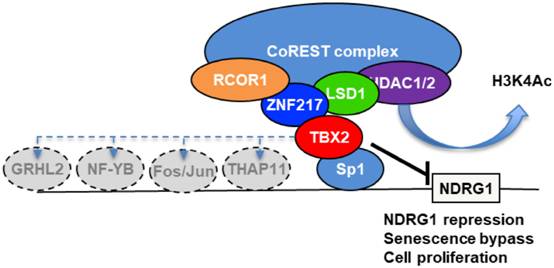
图8 TBX2-CoREST抑制NDRG1靶基因的机制。
参考文献
McIntyre AJ, Angel CZ, Smith JS, Templeman A, Beattie K, Beattie S, Ormrod A, Devlin E, McGreevy C, Bothwell C, Eddie SL, Buckley NE, Williams R, Mullan PB. TBX2 acts as a potent transcriptional silencer of tumour suppressor genes through interaction with the CoREST complex to sustain the proliferation of breast cancers. Nucleic Acids Res;50(11):6154-6173. doi: 10.1093/nar/gkac494.