






CircRNA DICAR——糖尿病心肌细胞焦亡的新型内源性调节因子
研究鉴定一个在糖尿病小鼠心脏中下调的保守的circRNA(circDICAR)。DICAR对糖尿病心肌病(diabetic cardiomyopathy,DCM)具有抑制作用,DICAR缺陷(DICAR+/-)小鼠表现出自发性心功能障碍、心肌细胞肥大和心脏纤维化,而DICAR过表达(DICARTg)小鼠DCM得到缓解。在细胞水平,作者发现过表达DICAR抑制糖尿病心肌细胞焦亡,而敲低DICAR增强糖尿病心肌细胞焦亡。在分子水平上,作者确定DICAR-VCP-Med12降解可能是DICAR介导效应的潜在分子机制。合成的DICAR junction part(DICAR-JP)表现出与整个DICAR类似的结果。此外,糖尿病患者循环血细胞和宣讲中DICAR的表达均低于健康对照组,这与糖尿病心脏中DICAR表达降低相一致。DICAR和合成的DICAR-JP可能是治疗DCM的候选药物。本研究于2023年3月发表于期刊《Signal Transduction and Targeted Therapy》上,IF:38.104。
技术路线:
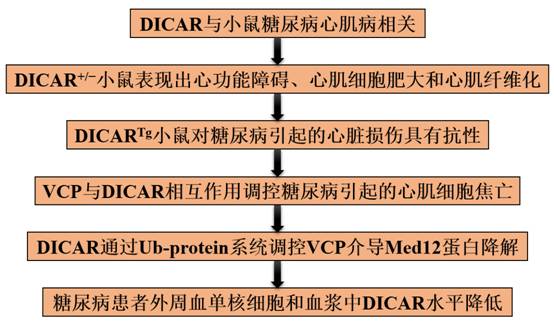
主要研究结果:
1、DICAR与小鼠糖尿病心肌病相关
作者通过circRNA微阵列筛选野生型(WT)小鼠和糖尿病db/db小鼠心肌组织中circRNA表达谱。结果发现,人和小鼠之间具有高度同源性的58个circRNAs在两组之间差异表达1.5倍以上(P<0.05)(GSE199133)。两组circRNA芯片代表性结果见图1a。选取5个差异表达倍数最高的circRNA进行RT-qPCR验证(图1b)。结果发现mm9_circ_008009是糖尿病心脏中表达下调最多的circRNA(图1b)。mm9_circ_008009抗核糖核酸酶R消化,而线性GAPDH mRNA易被降解(图1c)。根据circBase,mm9_circ_008009的亲本基因为Tulp4。mm9_circ_008009由Tulp4的exon1和intron形成(图1d)。mm9_circ_008009和hsa_circ_0131202在cDNA中均被扩增引物,而在gDNA中均未扩增(图1e-f),表明circRNA种类为环状。对PCR产物进行Sanger测序分析,发现mm9_circ_008009是通过“反向剪接”机制从Tulp4的外显子1和内含子产生的(图1g)。基于circBase,作者比对mm9_circ_008009与TULP4转录的36个人类circRNA之间的序列,发现hsa_circ_0131202与mm9_circ_008009之间的保守性为79.65 %。此外,鉴定hsa_circ_0131202的circRNA特征,在cDNA中用发散引物检测到该RNA,但在人心肌细胞(HCMs)的gDNA中没有检测到该RNA(图1f)。序列分析发现mm9_circ_008009结合位点约20 bp的片段与hsa_circ_0131202相似(图1g-h)。此外,hsa_circ_0131202的RNA模型结构与mm9_circ_008009的RNA模型结构相似(图1j-i),包含了反向剪接结(BSJ)。因此,来源相同、结构和序列相似的hsa_circ_0131202和mm9_circ_008009可能具有相同或相似的分子和生物学功能。荧光原位杂交实验(FISH)显示,mm9_circ_008009主要定位于心脏组织细胞中(图1k)。因此,作者将mm9_circ_008009和hsa_circ_0131202命名为糖尿病诱导循环相关circRNAs(DICAR)。
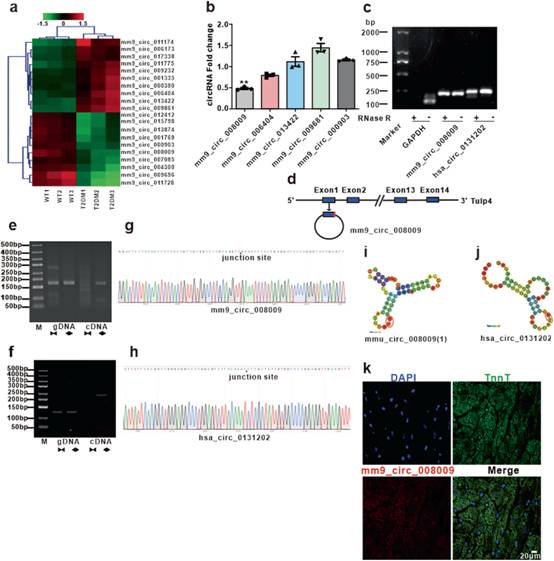
图1糖尿病性心肌病小鼠中circRNA的表达
2、DICAR+/−小鼠表现出心功能障碍、心肌细胞肥大和心肌纤维化
为测试DICAR在糖尿病性心肌病中的潜在作用,作者创造DICAR敲除小鼠。作者发现与对照小鼠相比,杂合子DICAR缺陷(DICAR+/-)小鼠的心功能受损。超声心动图检测结果显示,与同龄WT对照小鼠相比,DICAR+/-小鼠左心室收缩末期容积(LVESV)、左心室舒张末期容积(LVEDV)和左心室舒张末期内径(LVEDD)增加(图2a-b)。3月龄时,DICAR+/-小鼠的射血分数(EF)和短轴缩短率(FS)降低(图2a-b)。结果表明,DICAR表达降低会损害心脏功能,这与糖尿病患者的心脏功能障碍类似。
为检测DICAR在心脏重塑和纤维化中的潜在作用,作者分析DICAR +/-小鼠的心肌细胞面积和纤维化面积。与WT小鼠相比,年龄匹配的DICAR+/-小鼠wheat germ agglutinin(WGA)染色显示心肌细胞体积明显增大(图2c),Masson染色显示心肌间质胶原沉积明显增加(图2d),Ⅲ型胶原表达增强(图2e),反映心脏重构。
为检测DICAR对细胞焦亡的影响,作者在DICAR +/-小鼠心脏组织中检测一组细胞焦亡相关的生物标志物。本实验通过检测cleaved GSDMD、ASC、NLRP3、caspase-1 p21蛋白的表达来评估细胞焦亡的激活情况。结果显示,与WT对照组相比,DICAR +/-小鼠心脏组织中焦亡的所有蛋白标志物均上调(图2f)。
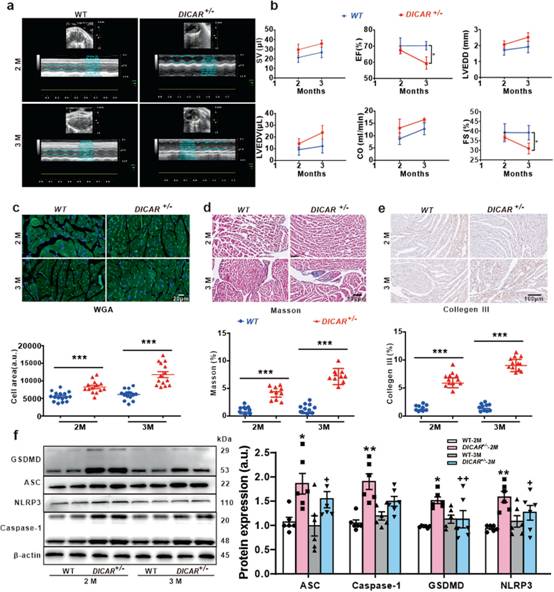
图2 DICAR对心功能及心肌重构的影响
3、DICARTg小鼠对糖尿病引起的心脏损伤具有抗性
为进一步研究DICAR在DCM发生发展中的作用,作者构建DICARTg小鼠,用于诱导2型糖尿病(diabetes mellitus type 2,T2DM),以探究其对DCM的潜在保护作用。对DICARTg小鼠进行超声心动图心功能评估。与非糖尿病野生型(WT)组相比,野生型糖尿病(WT-DM)组左室舒张末期内径(LVIDd)、左室收缩末期内径(LVIDs)、FS、SV、LVESV、LVEDV、CO等心功能指标均受损。可见,糖尿病DICARTg小鼠受损的心功能得到明显改善(图3a)。这些数据表明DICAR可能是DCM心脏保护中的关键circRNA。此外,与WT小鼠相比,WGA染色显示WT-DM小鼠心肌细胞体积增大,而糖尿病DICARTg小鼠心肌细胞体积增大被明显抑制(图3b)。在Masson染色心肌间质胶原沉积模式(图3c)和免疫组化Ⅲ型胶原表达模式(图3d)中,DICAR过表达也有类似的作用。这些数据表明DICAR可能是DCM过程中心脏保护的关键circRNA。
随后作者通过GSDMD、NLRP3、caspase-1和ASC的激活来确定心脏组织的焦亡。结果显示,与T2DM野生型对照小鼠相比,这些焦亡相关蛋白在T2DM DICARTg小鼠心脏组织中被显著抑制(图3e)。用ADDICAR(adenovirus-expressing DICAR expression)转染体外培养的小鼠心肌细胞48 h,再用AGEs(200 µg/mL)处理体外培养的小鼠心肌细胞48 h,过表达DICAR。Western blotting结果显示,在这些小鼠心肌细胞中,DICAR过表达抑制了AGEs对GSDMD、NLRP3、caspase-1和ASC活化的影响(图3f)。
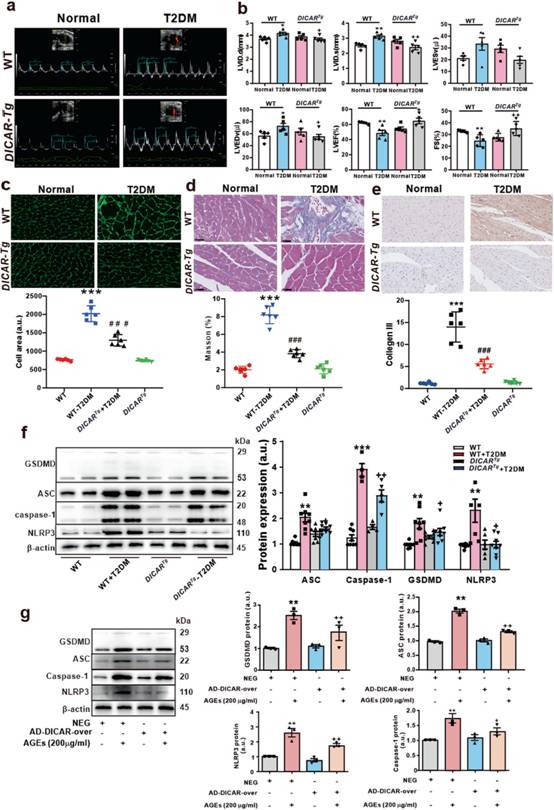
图3 DICAR功能的增加改善了DICARTg小鼠T2DM诱导的心功能
4、VCP与DICAR相互作用调控糖尿病引起的心肌细胞焦亡
对DICAR的第二个结构进行生物信息学分析,发现在连接结构域中存在一个特异的茎环结构(图1I)。由于作者发现DICAR定位于心肌细胞的细胞质中(图1g),因此作者确定DICAR是否可以与蛋白相互作用并调控蛋白。染色质分离RNA纯化-质谱(ChIRP-MS)是一种检测与ncRNA结合蛋白的方法,用于鉴定DICAR的结合蛋白。纯化DICAR结合位点的生物素标记探针,通过与小鼠心脏组织裂解液孵育进行circRNA pull - down实验,随后进行MS检测。共检测到50个蛋白,其中含缬酪肽蛋白(VCP)基于独特肽段和序列覆盖度得分最高。VCP,也被称为过渡内质网(tER)ATP酶或p97,是ATP酶家族中丰富且高度保守的成员,与多种细胞活动(AAA)相关,可控制泛素-蛋白酶体(Ub-Pr)降解途径中的关键步骤。平行反应监测(PRM)进一步验证23个DICAR结合的VCP蛋白,结果显示其在db/db小鼠心脏中的表达抑制高达31%(图4a)。但总VCP蛋白表达量在两组间无差异(图4b)。此外,为检测VCP与DICAR的结合能力,用VCP抗体进行RIP实验,然后对DICAR进行qPCR。如图4c所示,通过WB评估IP的特异性,发现在VCP IP中检测到独特的VCP条带,而在IgG IP中没有检测到。DICAR与VCP的结合量在DICARTg小鼠心脏中最高,在WT小鼠心脏中居中,在DICAR+/-小鼠心脏中检测不到。
为进一步探索DICAR-VCP结合模型,作者使用一个完整的基于结构的计算框架Rosetta-Vienna RNP-ΔΔG。如图4d所示,在对接区域,DICAR(19 bp)的二级RNA结构通过氢键、盐桥、氨基酸残基侧链网络和静电作用模型(附表S3)与VCP蛋白的氨基酸残基结合。此外,为验证DICAR与VCP的结合亲和力,作者合成了19 bp的DICARjunction part(DICAR-JP)。作者通过SPR测定DICAR和VCP之间的动力学速率常数。结果表明,DICAR-JP的结合常数(Kd)为5.4×10-9(图4e)。因此,作者证明DICAR可以通过连接片段与VCP结合,并且DICAR-JP是DICAR与VCP结合的关键片段。
为探讨DICAR-JP/VCP复合物在糖尿病诱导的心肌细胞焦亡中的作用,本研究体外合成了小鼠DICAR-JP(mDICAR-JP)、人DICAR-JP(hDICAR-JP)和VCP-siRNA。为探究DICAR-JP与VCP的联合作用,作者将DICAR-JP与VCP-siRNA共同转染MCMs。如图4f所示,DICAR-JP抑制AGEs诱导的细胞焦亡,VCP-siRNA增强DICAR-JP对细胞焦亡的抑制作用。因此,位于细胞质中的DICAR-JP/VCP复合物可能在DCM的细胞焦亡中发挥关键作用。
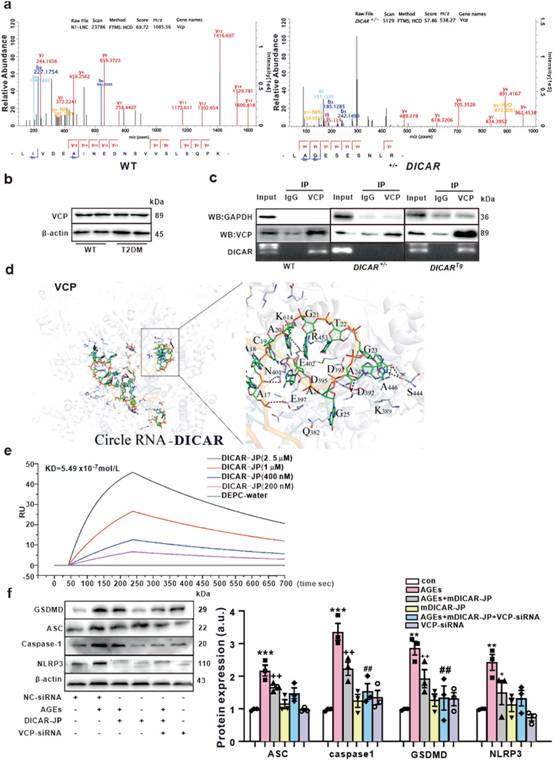
图4 VCP与DICAR在AGEs诱导心肌细胞焦亡中的相互作用
5、DICAR通过Ub-protein系统调控VCP介导Med12蛋白降解
为检测DICAR是否被定位于内质网的PE - DICAR探针标记,用ER - Tracker Green进行FISH检测(图5a)。作者还检验位于ER的VCP的DICAR调控。与WT组相比,DICAR+/-促进内质网中VCP的表达,DICARTg抑制内质网中的VCP(图5b)。
然而,过表达DICAR并不能使内质网中的VCP水平降低到正常生理条件下的水平。然后,作者检测DICAR在泛素化蛋白降解过程中是否通过与VCP相互作用介导蛋白的泛素化。在心脏中检测与VCP结合的蛋白质的泛素化水平。有趣的是,作者发现DICAR+/-小鼠的蛋白泛素化水平下调(图5c)。作者进一步证实DICAR结合位点的特殊序列是泛素化蛋白的分子伴侣,可以协助蛋白的泛素化降解。AGEs可下调DICAR,进而促进VCP在糖尿病心肌焦亡发生发展中的作用。DICAR的连接位点可能作为心肌细胞焦亡的关键调节因子。
基于VCP在蛋白降解中的功能及其与DICAR的相互作用,利用LC - MS检测WT和DICAR+/-小鼠心脏组织的蛋白水平(图5d)。如图5d所示,在泛素化过程中有5个蛋白表达下调,包括Myom1、Myh6、Med12、Myl2和Cavin2。作者随后检测这些蛋白在DICAR+/-小鼠心脏组织中的表达。如图5e所示,在这5个蛋白中,Med12是DICAR+/-小鼠心脏中表达下调最多的蛋白。为查清Med12 mRNA水平是否也受到调控,作者在DICAR+/-和DICARTg小鼠模型中检测了Med12的mRNA。如Fig 5f所示,Med12 mRNA无变化。在T2DM小鼠心脏中检测到这5个蛋白,其中Med12也是下调最多的蛋白(图5g)。结果提示,DICAR的下调可能通过Ub-protein降解系统解除VCP对Med12蛋白的降解作用。
Med12在T2DM心脏中表达降低,而在糖尿病DICARTg小鼠的心脏中表达上调(图5h)。mDICAR逆转了AGEs(200µg/mL, 24 h)对Med12蛋白的下调作用(图5i)。然而,与DICAR-JP相比,DICARJP和VCP-siRNA共转染并没有对逆转的Med12表达起到更好的作用(图5i)。这是VCP过表达质粒转染至细胞在探索DICAR-JPblock function的局限性。此外,Med12 siRNA单独也可诱导HL-1心肌细胞焦亡(图5j)。
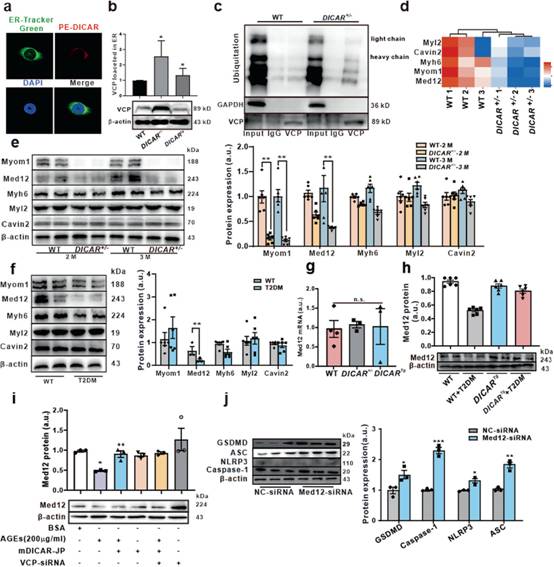
图5 VCP通过Ub-protein系统介导Med12蛋白降解
6、糖尿病患者外周血单核细胞和血浆中DICAR水平降低
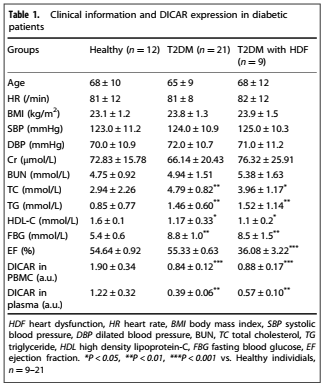
为了解DICAR表达与患者糖尿病之间的临床联系,作者测定3组年龄匹配的人外周血单核细胞(PBMCs)和循环血浆中的DICAR水平:正常健康对照组、糖尿病(T2DM)伴和不伴心功能不全患者。三组间性别、年龄、HR、BMI、SBP、DBP、Cr、BUN无统计学差异。糖尿病患者中TG、TC、HG较高(表1)。健康人PBMCs中DICAR水平为1.90±0.34,血浆中DICAR水平为1.22±0.32,糖尿病合并心功能不全患者(PBMCs : 0.84±0.12 ;血浆: 0.88±0.17)和糖尿病无心功能不全患者(PBMCs : 0.39±0.06 ;血浆: 0.57±0.10)中DICAR水平降低(表1)。
参考文献
Yuan Q, Sun Y, Yang F, Yan D, Shen M, Jin Z, Zhan L, Liu G, Yang L, Zhou Q, Yu Z, Zhou X, Yu Y, Xu Y, Wu Q, Luo J, Hu X, Zhang C. 2023. CircRNA DICAR as a novel endogenous regulator for diabetic cardiomyopathy and diabetic pyroptosis of cardiomyocytes. Signal Transduct Target Ther;8(1):99. doi: 10.1038/s41392-022-01306-2