






人胰岛的单核 RNA 测序识别新基因组 并区分具有动态转录组概况的 β 细胞亚群
该研究于2023年5月发表......
单细胞 RNA 测序 (scRNA-seq) 为人类胰岛细胞类型及其相应的稳定基因表达谱提供了宝贵的见解。然而,这种方法需要细胞解离,这使其在体内的应用变得复杂。另一方面,单核 RNA 测序 (snRNA-seq) 与冷冻样品兼容,消除了解离诱导的转录应激反应,并提供了可用于识别前体 mRNA 转录本的内含子序列的增强信息。作者使用snRNA-seq分析显示人胰岛内分泌细胞体外和体内差异选择性表达前四位的基因不是经典基因,而是一组新的非经典基因标记,包括ZNF385D、TRPM3、LRFN2、 PLUT(β细胞); PTPRT、FAP、PDK4、LOXL4(α-细胞); LRFN5、ADARB2、ERBB4、KCNT2(δ 细胞);和 CACNA2D3、THSD7A、CNTNAP5、RBFOX3(γ 细胞)。其次,通过整合来自人胰岛细胞的 scRNA-seq 和 snRNA-seq 的信息,我们区分了三个 β 细胞亚群:INS pre-mRNA 群 (β3)、中间 INS mRNA 群 (β2) 和 INS富含 mRNA 的簇 (β1)。这些显示不同的基因表达模式,代表体外和体内不同的生物动态状态。有趣的是,富含 INS mRNA 的簇 (β1) 成为体内主要的亚簇。综上所述,人胰岛细胞的 snRNA-seq 和 pre-mRNA 分析可以准确识别人胰岛细胞群、亚群及其体内动态转录组概况。
该研究于2023年5月发表发表在《Genomemedicine》,IF:15.266。
技术路线:

实验方法:人胰岛细胞和细胞核加工、人胰岛细胞移植、scRNA-Seq、snRNA-Seq、无监督数据分析、伪时间分析和RNA速度分析、通路分析、RNA原位杂交。
1、人胰岛细胞和细胞核的RNA-seq分析
图1A和B描述了用于这些研究的方法和数据分析工作流程。
作者分析了10,732个细胞和11,018个核。与scRNA-seq方法(图1C)相比,snRNA-seq方法中每个细胞的基因和读数测序的数量较低。在细胞核中测序的线粒体基因的TE百分比低于1%,明显低于在细胞制备中测序的线粒体基因(图1C)。在snRNA-Seq数据中,当分析内含子加外显子读取时,基因和读取的数量明显高于单独的外显子读取(图1D)。
基于上述数据质量,我们接下来将使用外显子读取的scRNA-seq与使用内含子和外显子读取的snRNA-seq进行比较,以改进基因检测和映射。scRNA-seq分析核转录本和细胞质转录本,其中大部分是细胞质转录本,而snRNA-seq分析的主要是核转录本,而在细胞核分离过程中,来自细胞质或粗糙ER的转录本最少。因此,我们预计,在scRNA和snRNA测序过程中,RNA-seq读数会不同。在细胞中,17±0.7%的读数为内含子读出,而在细胞核中为53±1.9%。另一方面,细胞中73±1.6%的读出为外显子读出,而细胞核中为32±0.8%。因此,只有当核与核或细胞与细胞进行比较时,基因表达相关性才会完全或接近完全线性(图1E),而当细胞与细胞核相比时,这种相关性较低(图1F)。
两种RNA测序方法(UMI>20)检测到的共同基因数量为16,452个(71.3%),而1433个基因(6.2%)仅在scRNA-seq中检测到,5184个基因(22.5%)仅在snRNA-seq中检测到(图1G)。表明两种方法检测到的几乎29%的基因对于相同的人胰岛样本是不同的,两种RNA测序方法可能揭示了胰岛细胞群体的同一性。
有趣的是,用Seurat的整合算法,使用外显子读数(scRNA-seq)或内含子加外显子读数(snRNA-seq)对细胞和细胞核进行无监督聚类,揭示了具有相似位置且在其UMAP中具有强烈重叠的簇(图1H,I)。

2、使用scRNA-seq和snRNA-seq对人类胰岛细胞类型进行监督分类
我们将scRNA-seq和snRNA-seq数据集投影到一个公开可用的Azimuth整合人类胰腺参考文献中,该参考文献包含六个不同的scRNA-seq数据集,这些数据集是使用Seurat的几种不同单细胞技术生成的(图1B和2A-D)。投影是通过scRNA-seq的外显子读数(图2A、B)和snRNA-seq的外显子或内含子加外显子读数(图2C、D)完成的。当我们将当前研究的人类胰岛scRNA-seq数据投影到参考上时,我们发现不同的人类胰岛细胞群几乎完美对齐,预测分数中位数为1,平均值为0.948(图2A,B)。使用外显子读数或内含子加外显子读数将snRNA-seq数据投影到参考也导致高度对齐(图2C,D),外显子加内含子的预测分数显着高于单独外显子(图2D)。为了确定观察到的与外显子加内含子读取的参考比对的更高概率是否可以用不同的UMI深度来解释,我们对包含内含子的库进行二次采样,中值为1247 UMI(外显子只读库的中值)并分析了参考对齐预测分数。我们发现在二次采样后,包含内含子的文库的比对预测分数仍然优于仅包含外显子的文库。这进一步验证了使用来自内含子和外显子读取的信息,并使用snRNA-seq对人类胰岛细胞群进行详细分析。数据还表明,来自scRNA-seq和snRNA-seq的人类胰岛细胞簇具有高度相似性,并且用于鉴定人类胰岛细胞类型簇时,包含内含子和外显子读数的snRNA-seq数据可与scRNA-seq数据可以互换。
接下来,我们测试了从scRNA-seq和snRNA-seq数据(图2A、C)生成的不同簇与不同内分泌细胞簇中典型基因的基因表达水平的关联(图2E-H)。在scRNA-seq中观察到已建立的典型基因细胞标记(GCG、INS、SST和PPY)与其他几种已知选择性标记与α-、β-、δ-和γ-细胞之间的强相关性。因此,在scRNA-seq分析中相应地分配了UMAP中的细胞类型(图2E,G)。然而,在scRNA-seq中观察到的这些典型基因标记的强相关性在snRNA-seq中对于α-、β-和γ-细胞较弱(图2F,H)。例如,在β细胞中,INS和IAPP是差异表达最多的基因,而在snRNA-seq数据集中它们不是差异表达最多的基因。这些结果强调需要鉴定新的基因组作为胰岛内分泌细胞的标记,以便在snRNA-seq分析中更恰当地定义它们。
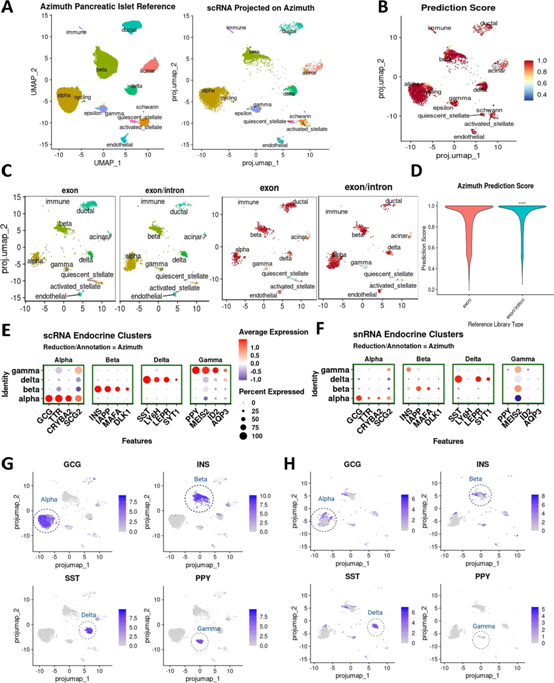
3、用于鉴定人胰岛内分泌细胞类型的snRNA-seq数据集中的新基因集
在scRNA-seq和snRNA-seq样本之间进行了差异基因表达分析,并研究了snRNA-seq富集基因的生物型。即使包含内含子读数,snRNA-seq中的大多数基因都是蛋白质编码基因(图3A)。接下来,注释了snRNA-seq数据,并投影到Azimuth的人类胰腺参考上,并用整个数据集测试了每个簇的差异基因表达(图1B)。使用这种方法,每个细胞簇中差异表达的基因被鉴定为p值~0和log2FC大于1.5。即使候选基因符合这些标准,我们也忽略了在其他细胞簇中表现出相当大的表达(log2FC>0.85)的基因。因此,制作了一个表,其中包含每种细胞类型的前四个差异表达基因(图3B)。有趣的是,scRNA-seq中定义内分泌细胞的前四个差异表达基因(即INS、GCG、STS或PPY)都没有出现在snRNA-seq中差异和选择性表达基因的简短列表中。这表明这些典型基因不是snRNA数据集中在α-、β-、δ-和γ细胞中差异表达最高的基因。为了确认这些新鉴定的内分泌细胞基因标记的可靠性,我们在snRNA-seq和scRNA-seq数据对象上测试了它们。它们在snRNA-seq和scRNA-seq的相应细胞簇中显示出清晰且主要是排他性的表达模式,但在snRNA-seq数据集中具有更高的表达(图3C,D)。
前几个内分泌细胞基因标记显示出比其相应的典型单细胞聚类基因标记更具特色的定位模式,分别用于snRNA-seq数据对象中的α-、β-、δ-和γ-细胞(图2H和3E)。值得注意的是,CNTNAP5在γ细胞中没有表现出高度不同的表达模式,但它被认为是基于高调整p值(1.58×10-5)和log2FC为1.654的γ细胞基因标记。有趣的是,在非内分泌细胞的snRNA-seq分析中,差异表达最高的基因包含在scRNA-seq中定义这些细胞类型的典型基因标记(REG1A、CFTR、FLT1、COL1A1和PRKG1,用于腺泡、导管、内皮、分别为激活的星状细胞、静止的星状细胞)。这表明人类内分泌细胞和非内分泌细胞之间关于规范基因的稳态转录本丰度的有趣二分法(图3F)。
为了验证这些来自snRNA-seq分析的新鉴定的基因作为细胞标记的存在,我们聚焦于β细胞,并使用RNA显微镜检测来自健康捐赠者的分散的人胰岛细胞中的ZNF385D mRNA。如图3G所示,ZNF385D mRNA在人β细胞中明显且唯一地被检测到。使用内含子探针的表达仅限于细胞核(图3G,上图),而使用外显子探针的信号定位在细胞质和细胞核中(图3G,下图)。
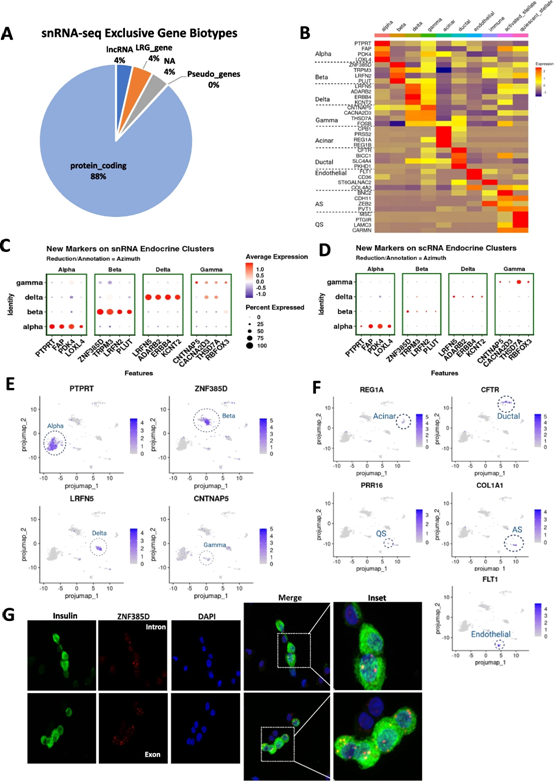
4、比较scRNA-seq和snRNA-seq分析在区分三种不同的β细胞亚型时的异同
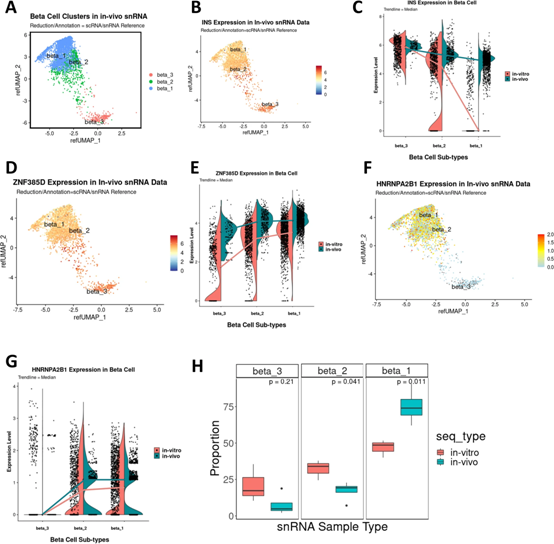
为了识别β细胞簇中的人类β细胞亚型,我们创建了一个新的数据对象,集成了scRNA-seq和snRNA-seq数据集,以比较INS基因表达模式(图1B和4A-C)。结合这两个数据集有效地增加了分析的能力,同时提供了来自细胞质和核转录组的额外信息。在对已识别的β细胞簇进行子集化后,我们生成了Louvain分辨率为0.8的簇(图4C)以分配三个β细胞亚簇。scRNA-和snRNA-seq数据对象之间的β细胞簇中的INS基因表达在拓扑位置方面是不同的(图4D、E)。聚类3在scRNA-seq数据中显示较低的INS表达,但在snRNA-seq数据对象中显示最高(图4E),这表明聚类3包括主要具有INS pre-mRNA的β细胞,因为snRNA-seq主要分析pre-mRNA。
接下来,我们创建了伪时间和RNA速度轨迹图,将聚类3作为基础(图4F),并根据伪时间或RNA速度轨迹重新排列了每个聚类的顺序,分为β1、β2和β3细胞(图4G)。使用两个数据集的综合数据进行伪时间或RNA速度分析时,从聚类3到聚类1的类似进展发生。单个scRNA-seq数据的RNA速度分析显示了从聚类3到聚类1的类似转变。有趣的是,仅使用snRNA-seq数据的RNA速度分析显示从聚类3末端到聚类3边缘的分叉,然后从聚类2到聚类1。这些结果表明,可以通过综合或单个RNA-seq数据观察到β细胞亚聚类的识别和转变,但是这种转变似乎在scRNA-seq和snRNA-seq之间有所不同。这很可能是因为RNA速度基于外显子-内含子读数比率,利用新转录的未剪接mRNA推断基因表达状态的时间导致,但是snRNA-seq中外显子-内含子读数较低,使得在聚类之间的转变的解释变得复杂。因此,对于综合或scRNA-seq数据,RNA速度是适当的,但是对于snRNA-seq,似乎RNA速度不太适合分析β细胞成熟的进展。
由于β1细胞簇中的细胞具有从scRNA-seq(成熟mRNA)β细胞簇对象推断出的稳定INS表达,但从snRNA-seq(pre-mRNA)β细胞簇对象推断出非常低的INS表达,我们将这些细胞视为富含INS的细胞亚群。接下来,我们观察了ZNF385D表达,它是β细胞中snRNA-seq中差异表达最高的基因,发现scRNA-seq数据对象中β细胞亚群中的表达最小,但snRNA-seq数据中的拓扑位置不同对象(图4H,I),其中β1代表具有高ZNF385Dpre-mRNA表达的细胞,与INS表达相反(图4E)。接下来,我们观察了INSmRNA结合蛋白HNRNPA2B1的表达,这是一种调节INSmRNA稳定性和翻译的RNA结合蛋白,以确定β2细胞簇是否代表β3和β1之间的过渡阶段。如图4J、K所示,与聚类3相比,聚类2和聚类1中的HNRNPA2B1表达增加,表明聚类2可能代表“转变中”β细胞类型,其中细胞从转录活性β3细胞转变为β1细胞具有成熟储存的INS mRNA。
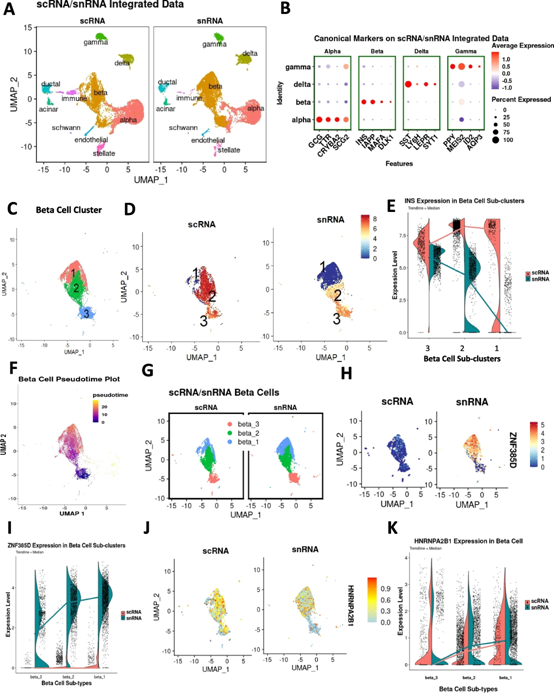
5、三种不同β细胞亚型的基因通路分析
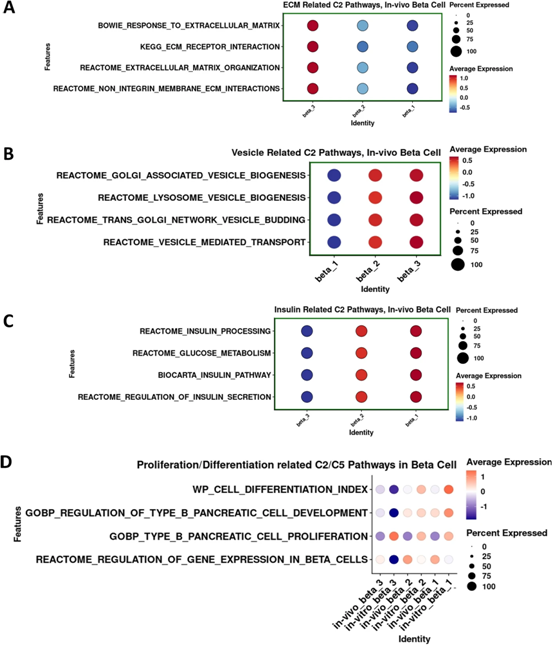
为了研究β3、β2和β1之间潜在的生物学差异,我们对来自scRNA和snRNA-seq实验的组合数据集进行了GSEA。有趣的是,定义细胞外基质(ECM)形成、相互作用和反应的生物过程的基因富集独特地存在于β3亚群中(图5A)。同样,GSEA也表明细胞内囊泡出芽、运输和形成的生物学过程主要存在于β2亚群中(图5B),而胰岛素分泌、加工和葡萄糖代谢生物学过程的基因主要存在于β2亚群中。在β2和β1子簇中表示(图5C)。当我们将来自两种RNA-seq方法的数据集输入GSEA时,还获得了关于对β细胞的发育、分化、基因表达调控和增殖具有重要意义的几个生物学过程的附加信息。
如图5D所示,参与β细胞增殖的基因在β3亚群中表达更高,参与基因表达调控的基因在β2亚群中突出,而参与β-细胞发育和分化在β1亚群中表达更高。这些结果清楚地描述了不同β细胞亚群中特定细胞功能的不同基因集,强调了人类胰岛中β细胞的异质性。
6、人体内胰岛的snRNA-seq分析
接下来,我们试图使用移植到血糖正常的免疫抑制小鼠体内的人类胰岛移植物来检查体内不同的人类胰岛细胞群。为此,我们将来自四名健康供体的1000个人类IEQ移植到RAG1-/-小鼠的肾囊中,并在移植后3个月收获移植物(图6A)。
通过Minute™单核分离试剂盒直接从移植物中提取细胞核,在质量评估和计数后将每个样本5000-16,000个细胞核加载到10X GenomicsChromiumController中,poly-A转录本逆转录扩增,cDNA标记,以及得到的文库测序深度为每个样本250-5亿个读数。最终,分析了7765个细胞核。基因数为2226±263,基因计数为4125.64±263,决定测序效率的可用读数与测序读数之比为0.999±0.001。在细胞核制备物中测序的线粒体基因百分比低于1%。snRNA-seq数据被投射到Azimuth人类胰腺参考上,以使用规范基因集以及之前描述的新基因集(图1A和6A)识别胰岛细胞群。此外,snRNA-seq数据被投影到来自体外研究的集成scRNA-seq和snRNA-seq数据上,用于分析不同的β细胞亚群(图6A)。去除基因计数和/或UMI计数异常值或线粒体基因高表达或环境RNA污染≥20%的细胞。在质量控制过程中,我们发现≤10%的小鼠基因比率对于在这种胰岛移植环境中识别体内不同细胞类型是最佳的,而小鼠基因对聚类模式没有显着影响。
使用Azimuth的基于参考的缩减和注释,我们确认了七个不同的人类胰岛细胞簇,主要由内分泌细胞组成(图6B)。为简单起见,我们从数据集中省略了少于五个细胞的簇。通过体外snRNA-seq分析鉴定的基因标记集(图3C)在基于平均表达的点图(图6C)和散点图(图6D)与典型基因标记的非特异性模式相比。当使用基于内部参考的减少和注释时,体外snRNA-seq分析中新鉴定的基因集的这种特异性细胞簇比对持续存在(图6E-G)。
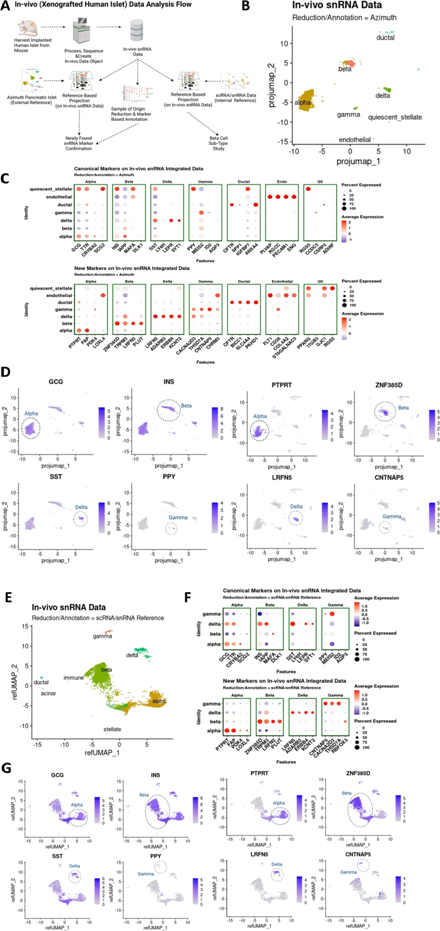
7、人体内胰岛中的β细胞亚型
首先,我们确定在scRNA-seq/snRNA-seq体外研究中鉴定的β细胞亚型是否在体内环境中仍然存在于人类胰岛中。因此,我们将体内snRNA-seq数据集投影到体外scRNA-/snRNA-seq数据集参考上,该数据集预先标记了β细胞亚型(图6E)并提取了β细胞簇(图7A)。我们从主要数据簇中分离出β细胞亚簇并检查基因表达模式(图7A-H)。有趣的是,与体外人胰岛的snRNAseq数据相反,体内人胰岛中的β1簇与β2和β3簇相比显示出相似水平的INS表达(图7B,C)。当分析先前来自人类胰岛移植物的snRNA-seq数据集时,观察到类似的结果。此外,在我们的数据集中,ZNF385D和HNRNPA2B1的表达在体内和体外人胰岛的β细胞亚群中似乎相似(图7D-G)。重要的是,与体外相比,体内人胰岛中β1亚群中具有稳定INS表达的细胞比例显着增加,而β2转换群和主要含有INS pre-mRNA的β3细胞比例显着降低(图7H)。我们推测这可能代表细胞从较低的INS表达β3和β2组到更成熟的INSmRNAβ1组的表型或成熟转移,与生理性较低的体外条件相比,可能反映了体内微环境的成熟特征。
我们还使用与上述相同的方法对体内人类胰岛的不同β细胞亚群进行了基因集富集分析(图5和图8)。正如在体外观察到的(图5),与β2和β1细胞亚型相比,β3细胞亚型显示出更高水平的参与ECM生物过程的基因表达(图8A)。在β2和β1细胞亚型中,参与细胞内囊泡出芽、运输和形成、胰岛素分泌、加工和葡萄糖代谢、β细胞分化、发育、增殖和基因表达调控等生物学过程的基因表达在体内和体外保持相当(图8B-D)。值得注意的是,人类β3细胞体内与体外β细胞增殖相关的基因表达低于体外(图8B-D),再次表明体内微环境可能为β细胞提供线索以支持功能更强、但与生理性较低的体外环境相比,有丝分裂状态较低。
参考文献:
Kang,R.B.,Li,Y.,Rosselot,C.etal.Single-nucleusRNAsequencingofhumanpancreaticisletsidentifiesnovelgenesetsanddistinguishesβ-cellsubpopulationswithdynamictranscriptomeprofiles.GenomeMed15,30(2023).https://doi.org/10.1186/s13073-023-01179-2