






糖尿病伤口愈合障碍新机制:中性粒细胞胞外诱捕网(NETs)通过内质网应激诱发成纤维细胞铁死亡
糖尿病患者伤口延迟愈合显著增加截肢和全身感染的风险。然而,其潜在的分子机制尚不清楚。成纤维细胞通过分泌胶原蛋白参与创面修复,其功能障碍会严重影响创面愈合和组织再生。越来越多的证据表明,中性粒细胞胞外诱捕网(NETs)的过度产生会加剧局部炎症并诱导细胞死亡,从而延迟伤口愈合。尽管有这些发现,NETs导致伤口愈合障碍的确切机制仍不清楚。本研究利用体外和体内模型来研究NETs通过内质网应激促进成纤维细胞铁死亡并促进受损糖尿病伤口愈合的分子机制。我们的研究结果表明,NETs诱导成纤维细胞的铁死亡,从而损害其胶原分泌能力。我们进一步证明了IRE1α/XBP1,一个关键的内质网应激通路,参与了NETs诱导的成纤维细胞铁死亡。此外,靶向该通路可显著促进糖尿病创面愈合。这些发现为治疗难治性糖尿病创面提供了新的分子靶点和潜在的治疗策略。该研究于2025年5月发表在《Free Radical Biology and Medicine》,IF:8.2。
技术路线
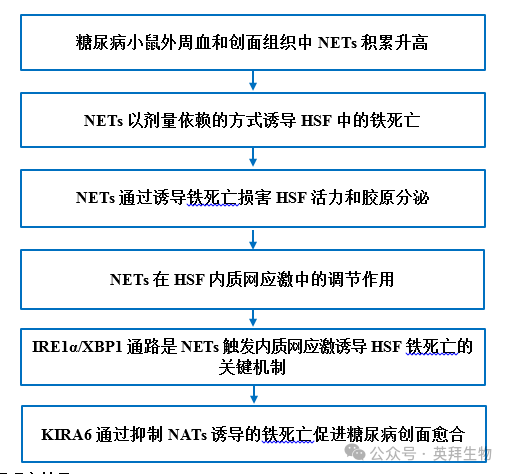
主要研究结果:
1.糖尿病小鼠外周血和创面组织中NETs积累升高
为研究NETs在糖尿病创面愈合中的作用,我们建立了糖尿病小鼠模型,并评估了NETs生物标志物CitH3和MPO在创面组织和外周血中的表达水平。代表性免疫荧光图像显示,与对照组小鼠相比,糖尿病小鼠创面皮肤中CitH3和MPO的浸润显著增加(图1A)。对外周血中NETs水平的分析表明,糖尿病小鼠中的CitH3和MPO水平显著高于健康对照小鼠(图1B和C)。这些结果表明,在糖尿病条件下,外周血和创面组织中存在广泛的NETs蓄积,表明NETs在糖尿病慢性创面的发病机制中是必不可少的。
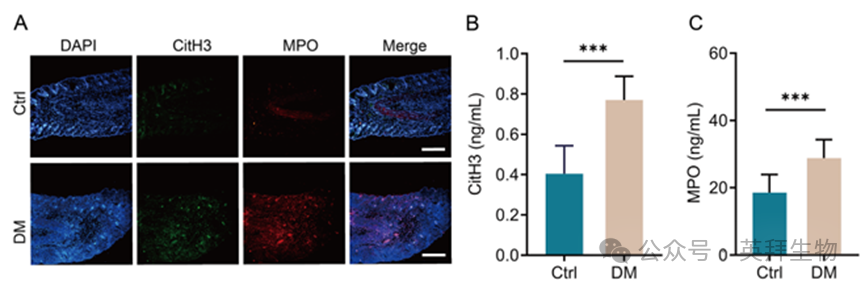
图1.糖尿病小鼠伤口组织和外周血中NETs积累升高
2.NETs以剂量依赖的方式诱导人皮肤成纤维细胞(HSF)中的铁死亡
在本文中,我们使用中性粒细胞分离试剂盒从健康志愿者的外周血中分离中性粒细胞,并将其与100 nM PMA孵育4 h以诱导NETs形成(图2A)。随后使用免疫荧光观察NETs的形成(图2B)。我们利用微阵列基因表达谱进行了转录组学分析,以检测NETs对成纤维细胞功能的影响及其潜在的调控机制。火山图显示,与对照组相比,NETs处理组有657个上调和371个下调的DEGs(图2C)。热图分析显示,PTGS2是NETs处理后上调最显著的基因之一,表明其在铁死亡调节中的潜在作用(图2D)。为验证这些转录组学发现,我们评估了用逐渐增加浓度的NETs(0、25、50和100 μg/mL)处理24 h的HSF细胞中PTGS2和SLC7A11(铁死亡的关键调节因子)的mRNA水平。此外,qPCR显示PTGS2 mRNA水平呈剂量依赖性上调(图2E),而SLC7A11 mRNA表达随NETs浓度的增加而显著下调(图2F)。NETs处理以浓度依赖性方式显著降低HSF活力(图2G)。NETs暴露后LPO水平显著升高(图2H),而GSH水平以剂量依赖性方式降低(图2I)。透射电镜显示经nets处理的HSF中存在严重的线粒体损伤,包括线粒体萎缩、嵴断裂和膜完整性受损(图2J)。WB分析证实,NETs处理后PTGS2表达显著上调,而SLC7A11水平降低(图2K-M)。此外,关键ECM蛋白,COL I和COL III,以剂量依赖性方式显著下调(图2N-P)。这些结果表明,NETs以剂量依赖性方式诱导HSF发生铁死亡,损害其胶原分泌能力。
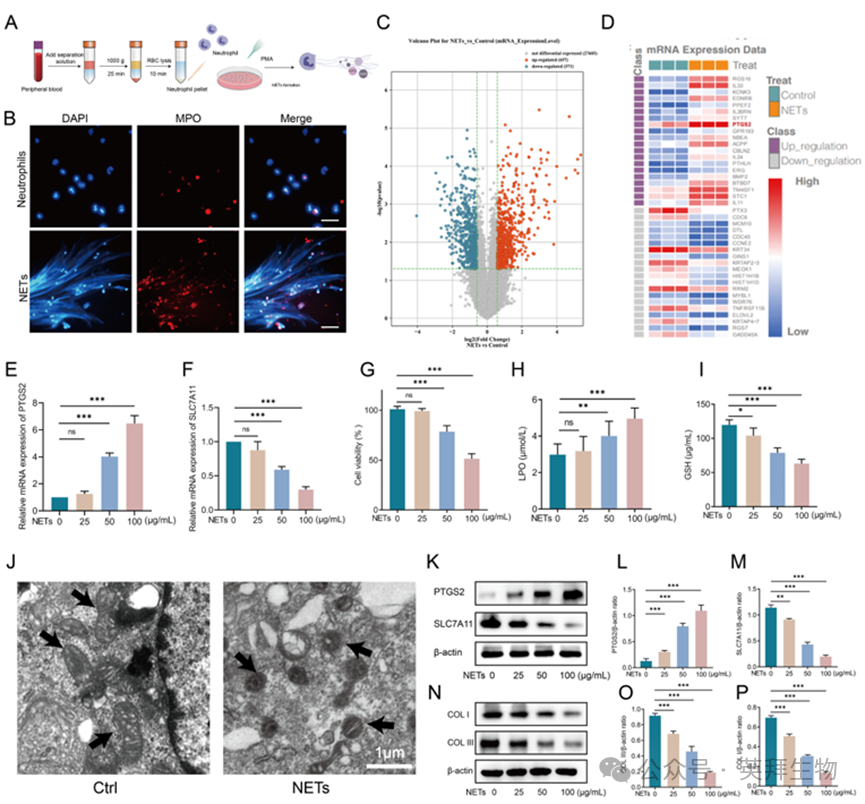
图2.NETs可诱导HSF发生铁死亡
3. NETs通过诱导铁死亡损害HSF活力和胶原分泌
为探讨铁死亡在NETs诱导的细胞功能障碍中的作用,我们研究了铁死亡抑制剂ferrostatin-1 (Fer-1)对NETs诱导的HSF细胞功能障碍的影响。我们发现,NETs显著增加细胞内Fe2+水平,与铁死亡诱导剂Erastin相当,而Fer-1预处理有效地缓解了这一效应(图3A)。同样,Fer-1预处理显著减少了ROS的积累(图3B)。NETs处理显著增加了PTGS2和ACSL4的表达,同时降低了COL I、COL III、GPX4和SLC7A11(图3C-I)。Fer-1有效地逆转了这些作用。使用C11 LPO探针的流式细胞术(图3J)以及LPO和谷胱甘肽GSH水平的测定(图3K和L)进一步证实,NETs在HSF中诱导LPO。正如预期的那样,Fer-1有效地减轻了NETs对HSF的影响。Fer-1预处理减轻了NETs诱导的细胞毒性,这可以通过提高细胞活力得到证明(图3M)。这些结果表明,NETs通过增加LPO和降低抗氧化能力促进成纤维细胞铁死亡,可能导致细胞死亡和功能障碍。
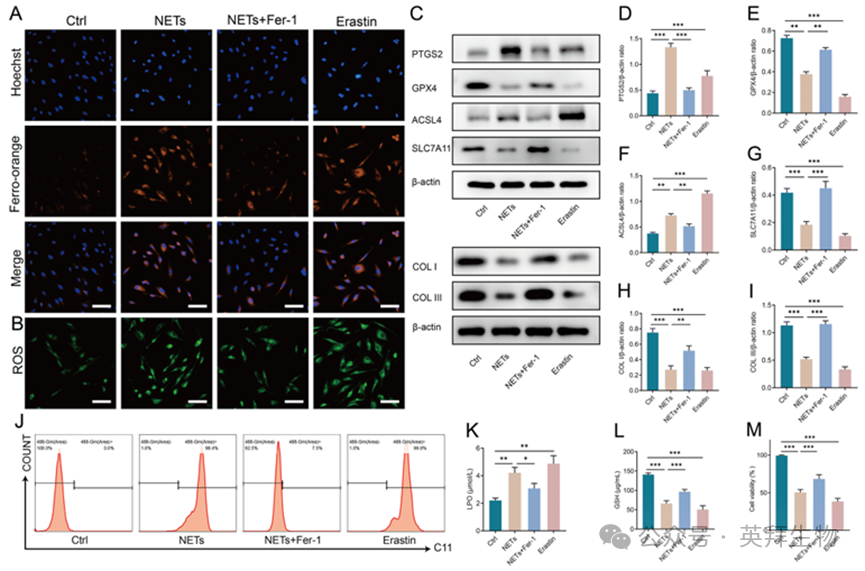
图3.抑制铁死亡可减轻NETs诱导的HSF功能障碍并恢复细胞活力
4. NETs在HSF内质网应激中的调节作用
内质网应激是细胞内ROS和LPO积累的重要调控途径。因此,我们假设NETs通过激活内质网应激来调节HSF的铁死亡。透射电镜分析显示,与对照组相比,NETs处理组线粒体出现铁死亡(图4A,黑色箭头),同时粗面内质网显著扩张(图4A,白色箭头),表明内质网应激的激活。我们进一步分析了三个主要的内质网应激感受器:ATF6, IRE1α和PERK的激活。WB分析显示,与对照组相比,NETs处理显著增加了IRE1α和PERK的磷酸化,而ATF6的表达没有变化(图4B-E)。这些发现表明,NETs主要通过IRE1α和PERK途径而不是ATF6途径诱导内质网应激。
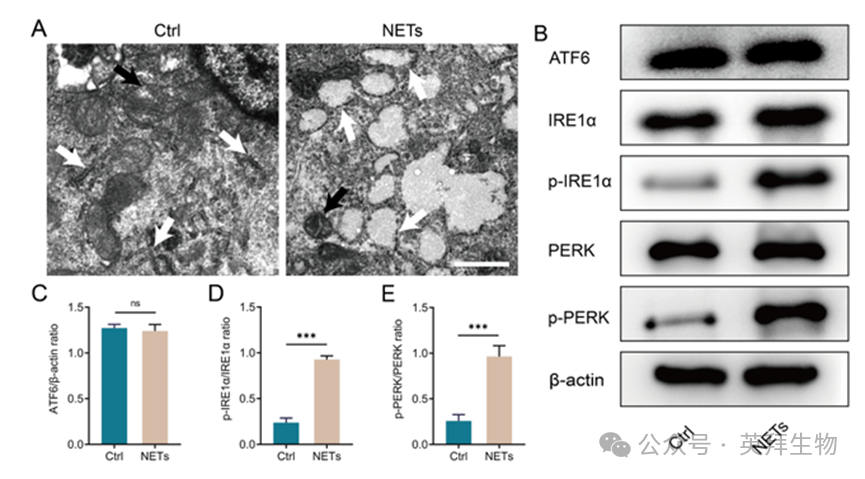
图4.NETs在体外激活内质网应激
5.IRE1α/XBP1通路是NETs触发内质网应激诱导HSF铁死亡的关键机制
为进一步研究IRE1α和PERK通路在铁死亡中的调控作用,我们用IRE1α抑制剂(KIRA6, 10 μM)、PERK抑制剂(GSK2606414, 2 μM)和铁死亡抑制剂(Fer-1, 15 μM)预处理HSF,然后用NETs处理24 h。采用ER-Tracker评估内质网扩张情况。荧光图像显示,经NETs处理的HSF中,ER荧光强度明显增强,而经KIRA6或GSK2606414预处理后,ER荧光强度减弱。相反,Fer-1预处理没有显著改变NETs处理的HSF中的ER荧光强度(图5A)。NETs + KIRA6组ROS水平明显降低,细胞存活率升高。GSK2606414对ROS积累的抑制作用不如KIRA6显著(图5B),对细胞活力的影响最小(图5C)。这些结果表明,抑制铁死亡不影响NETs诱导的内质网扩张。PERK通路对NETs诱导的脂质过氧化的调节作用很小。WB分析显示,KIRA6预处理的HSF中,PTGS2表达降低;而SLC7A11表达增加。GSK2606414预处理的NETs组中PTGS2和SLC7A11的表达水平无明显变化。Fer -1预处理的NETs组中IRE1α和PERK表达水平保持不变(图5D-H)。这些结果表明,NETs主要通过IRE1α通路的激活来调节内质网应激,从而影响HSF的铁死亡。虽然NETs激活了PERK通路,但它们对HSF的铁死亡没有显著的调节作用。Fer-1对内质网扩张和内质网应激通路蛋白的表达无显著影响,表明NETs诱导的HSF铁死亡主要受IRE1α通路的调控。
为研究NETs是否通过激活IRE1α/XBP1通路促进HSF的铁死亡,我们使用FerroOrang染色检测细胞内Fe2+的积累。FerroOrange的荧光图像显示,KIRA6可以抑制HSF中NETs诱导的细胞内Fe2+的积累(图5I)。WB分析显示,NETs刺激显著上调了HSF中XBP1和CHOP的表达,表明内质网应激的IRE1α/XBP1轴被激活。KIRA6预处理可抑制ACSL4的表达,同时恢复GPX4的表达(图5J-N)。透射电镜显示NETs处理后细胞内质网和线粒体超微结构发生明显改变。与NETs处理组相比,KIRA6预处理减轻了HSF的内质网扩张,并显著恢复了线粒体形态和超微结构(图5O)。流式细胞术分析显示,KIRA6抑制了NETs诱导的HSF脂质过氧化(图5P)。我们的研究结果表明,NETs通过激活IRE1α/XBP1通路触发HSF中内质网应激介导的铁死亡。
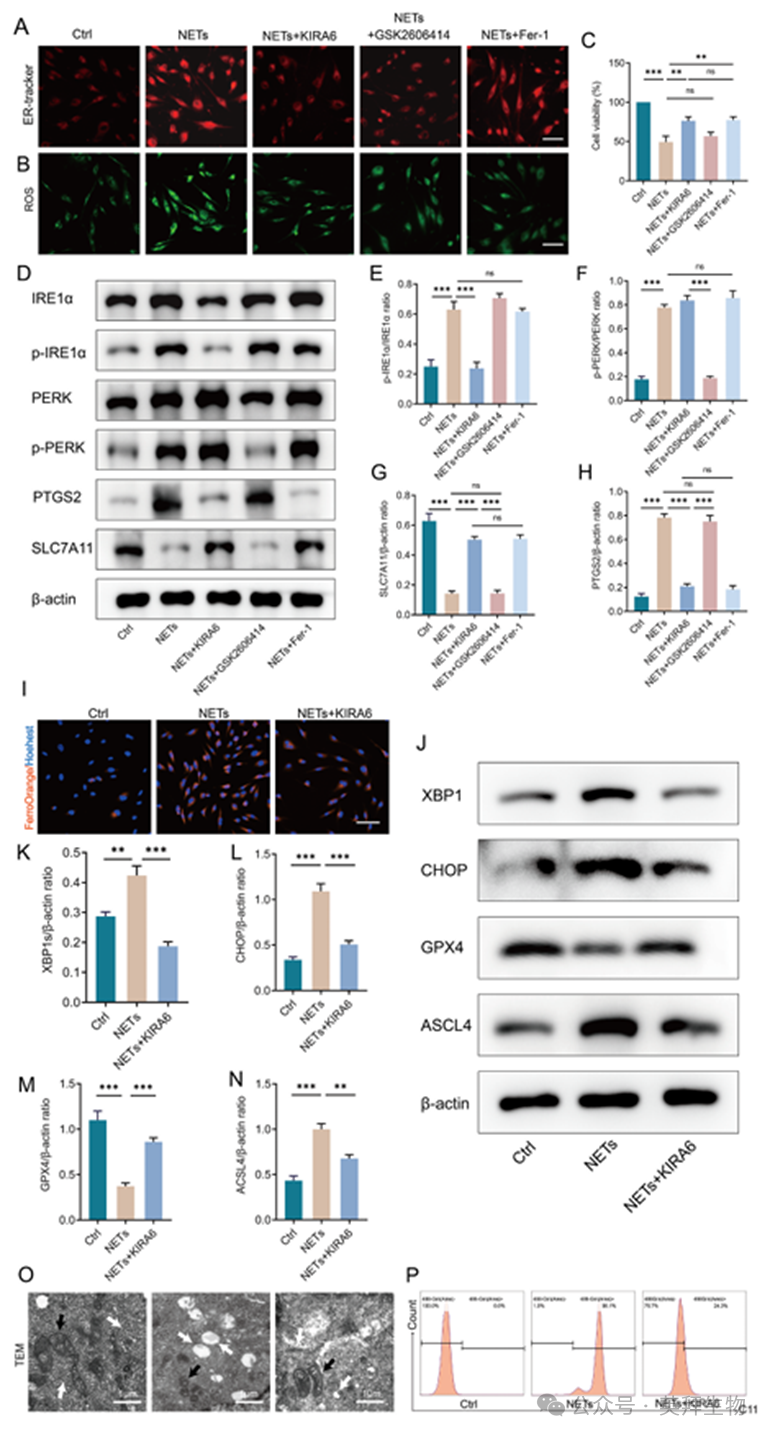
图5.IRE1α/XBP1通路是NETs触发内质网应激诱导HSF铁死亡的关键介质
6.KIRA6通过抑制NETs诱导的铁死亡促进糖尿病创面愈合
我们创建了糖尿病小鼠伤口愈合模型,并在14天内监测伤口愈合情况,以评估NETs诱导的铁死亡对伤口愈合的影响(图6A)。不同时间点(0、3、7和14天)的代表性伤口图像表明,与未经治疗的糖尿病小鼠相比,DM + KIRA6组小鼠的伤口闭合改善(图6B和C)。此外,KIRA6处理显著增强了皮肤创面ⅰ型胶原的表达,恢复了皮肤的胶原分泌能力。此外,内质网应激相关标志物(IRE1α, XBP1和CHOP)的表达下降,伴随脂质过氧化相关蛋白PTGS2和ACSL4的下调和抗氧化蛋白GPX4的上调(图6D)。这些结果表明,KIRA6通过抑制NETs诱导的铁死亡来减轻糖尿病慢性创面。
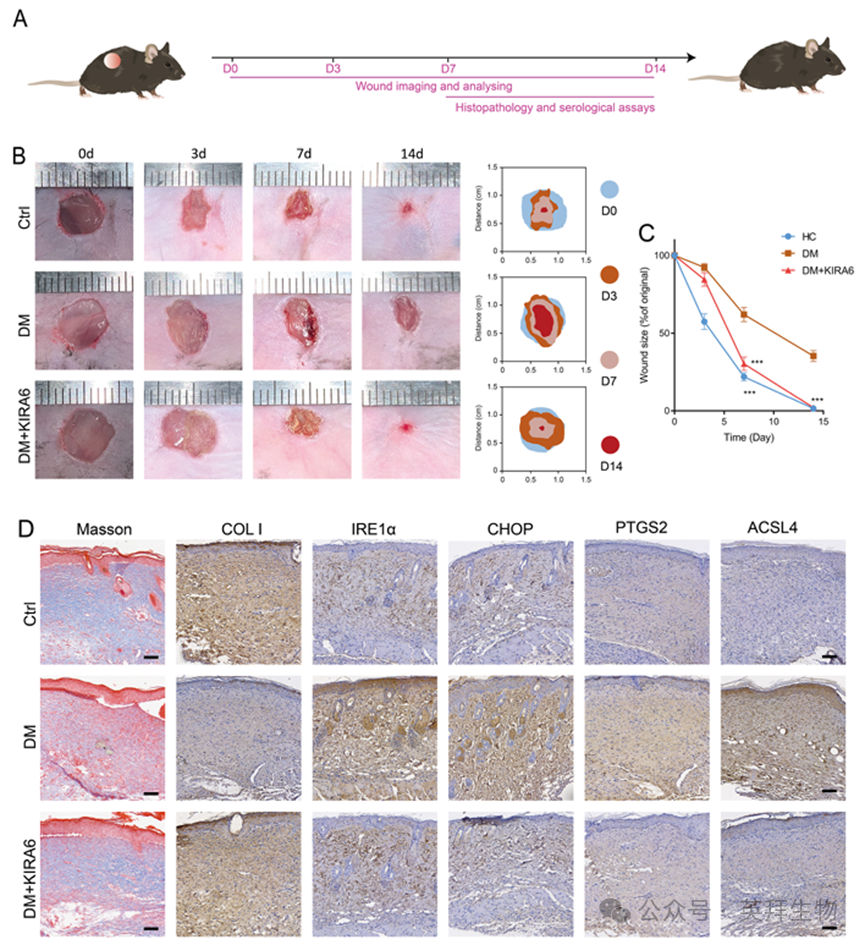
图6.KIRA6通过抑制铁死亡促进体内糖尿病创面愈合
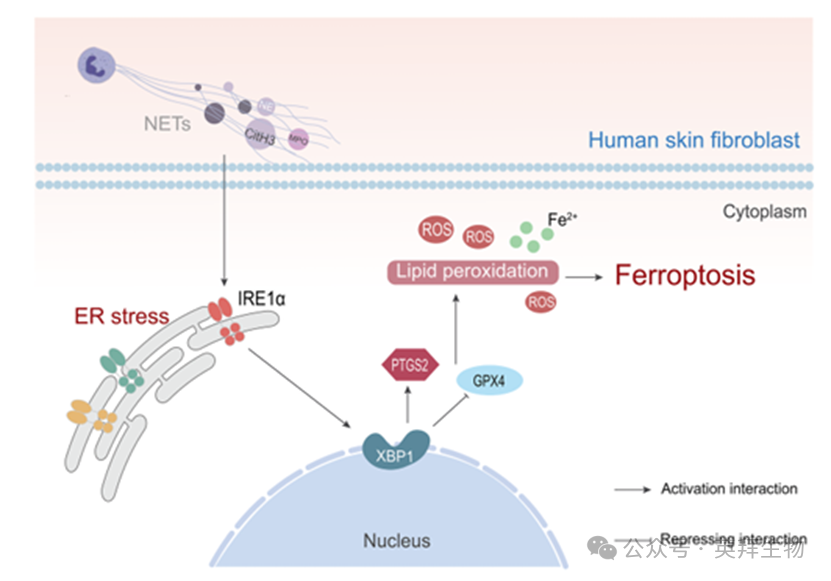
图7.示意图总结。NETs通过IRE1α/XBP1通路加剧脂质过氧化和ROS释放,从而诱导HSF发生铁死亡。
结论
综上所述,我们通过体内和体外模型证明了NETs在糖尿病伤口延迟愈合中的作用。具体而言,NETs通过激活IRE1α/XBP1信号通路启动内质网应激,导致HSF发生铁死亡,损害成纤维细胞的修复功能(图7)。此外,KIRA6治疗有效地减轻了NETs诱导的细胞损伤,并显著促进伤口愈合。调节NETs触发内质网应激诱导细胞死亡的分子机制是缓解糖尿病伤口延迟愈合的一种有前景的治疗策略。
参考文献
Zhao H, Liu Y. Neutrophil extracellular traps induce fibroblast ferroptosis via IRE1α/XBP1-mediated ER stress to impair diabetic wound healing. Free Radic Biol Med. 2025 Aug 16;236:17-27. doi: 10.1016/j.freeradbiomed.2025.05.391. Epub 2025 May 14. PMID: 40379156.