






Serpina3k 乳酸化可预防心脏缺血再灌注损伤
乳酸在缺血再灌注损伤期间产生,已知可促进蛋白质的乳酸化,其作用存在争议。通过质谱分析小鼠心肌在缺血再灌注损伤期间的乳酸化组和蛋白质组,作者发现Serpina3k蛋白表达及其在赖氨酸351位点的乳酸化在再灌注时均增加。Serpina3k及其人类同源物SERPINA3主要在心脏成纤维细胞中表达,而不是在心肌细胞中。生化分析表明,Serpina3k的乳酸化增强了蛋白质的稳定性。通过使用Serpina3k基因敲除小鼠和过表达乳酸化缺陷突变体的小鼠,作者发现Serpina3k以赖氨酸351位点乳酸化依赖的方式保护心脏免受损伤。机制上,缺血再灌注刺激的成纤维细胞分泌Serpina3k/SERPINA3,并通过旁分泌方式保护心肌细胞免受再灌注诱导的凋亡,部分通过激活心脏保护性再灌注损伤挽救激酶(RISK)和存活因子增强(SAFE)通路。作者的结果揭示了蛋白质乳酸化在心脏缺血再灌注损伤中的关键作用,这可能为治疗提供新的思路。该研究于2025年1月发表于《Nature Communication》,影响因子15.7。
技术路线

研究思路
1.全局乳酸化组分析心肌缺血/再灌注损伤
为了全面表征梗死和再灌注心脏中蛋白质赖氨酸乳酸化(Kla)的状态,作者采用了一种强大的四维(4D)无标记定量方法进行全球乳酸化组和蛋白质组分析(图1a)。4D蛋白质组学整合了离子迁移率这一第四维度,提高了肽段和蛋白质的鉴定能力。9周龄小鼠分别接受假手术(Sham)、心肌梗死(MI)30分钟或心脏缺血/再灌注(I/R)6小时处理。选择这些时间点是基于先前的MI研究以及支持在刺激后12小时内分析蛋白质乳酸化的报告。在乳酸化组分析中,作者共鉴定出1674个Kla位点,涉及380种蛋白质,其中1472个Kla位点来自333种蛋白质被定量(补充表S1;补充数据1和2)。蛋白质组分析鉴定出3279种蛋白质,其中2759种在严格过滤后被定量(补充表S1;补充数据3)。乳酸化组和蛋白质组数据均通过将修饰肽段的相对定量值除以相应蛋白质的相对定量值进行归一化处理。在乳酸化组和蛋白质组分析中,大多数肽段由7-20个氨基酸组成,符合质量控制要求(补充图1a,b)。主成分分析显示,I/R组的乳酸化组和蛋白质组与Sham和MI组存在偏离,表明在这一阶段发生了更大的变化(补充图1c,d)。皮尔逊相关性分析显示,Sham和MI组之间相似度更大,而I/R样本在乳酸化组和蛋白质组中均显示出与MI和Sham更明显的偏离(补充图1e)。乳酸化组的相对标准偏差高于蛋白质组,但均低于30%的接受阈值(补充图1f,g)。为了研究乳酸化位点附近的氨基酸模式,作者使用MEME套件中的MoMo工具(motif-x算法)来揭示鉴定的乳酸化位点附近的过代表模式。发现丙氨酸(A)在-4位(xxxxxxAxxx_K_xxxxxxxxxx)和赖氨酸(K)在+7位(xxxxxxxxxx_K_xxxxxxKxxx)是乳酸化的显著过代表模式(模式得分分别为7.77和6.87)(补充表S2;补充数据4)。通过热图可视化乳酸化位点附近的氨基酸频率。赖氨酸(K)残基在-10和+7位富集,丙氨酸(A)残基在-6、-4和+4位富集,甘氨酸(G)在-1位富集(补充图1h;补充数据5)。作者还使用iceLogo调查乳酸化位点周围的氨基酸,观察到类似模式(补充图1i;补充数据6)。为了确定乳酸化是否对特定蛋白质结构特征有偏好,作者使用NetSurfP计算了乳酸化在α-螺旋、β-折叠和卷曲中的概率,但未发现偏好,表明蛋白质的二级结构对目标赖氨酸残基的乳酸化没有影响(补充图1j;补充数据7)。相应地,所有赖氨酸残基和乳酸化赖氨酸的表面可及性几乎相同。
作者进一步检查了MI和I/R期间全球Kla分布的变化,分别与Sham进行比较。在MI中,约2.3%的蛋白质表现出乳酸化上调,而仅有约0.5%显示出乳酸化下调。类似地,在I/R中,约4.3%的蛋白质显示出乳酸化上调,而仅有约0.3%显示出乳酸化下调(图1b;补充数据8)。这些结果表明,MI和I/R主要诱导蛋白质乳酸化。蛋白质组的变化与乳酸化组相似,与下调的蛋白质相比,MI和I/R中上调的蛋白质更多(图1c;补充数据9)。采用更严格的截止值(log2比值=1.3),作者进一步定量了MI和I/R中Kla位点和蛋白质的变化数量。在MI中,有25个上调和2个下调的Kla位点,分别对应18和2种蛋白质的乳酸化上调和下调。相比之下,在I/R中,有71个上调和2个下调的Kla位点,分别对应41和2种蛋白质的乳酸化上调和下调,表明再灌注后乳酸化的持续影响(图1d;补充数据10)。在蛋白质组分析中,MI和I/R组分别有19和111种上调的蛋白质(图1e;补充数据11)。I/R组还有额外的26种下调的蛋白质(图1e)。
接下来,作者试图研究MI和I/R期间显著差异乳酸化和差异表达的蛋白质,并进行基因本体(GO)分析以了解其功能相关性。乳酸化蛋白被分为三个不同的簇,基于它们随时间的乳酸化状态(图1f;补充数据12)。第一组蛋白(乳酸化组C1)在MI期间几乎没有增加,随后在I/R时急剧上升(补充图2a)。许多与细胞呼吸(如Mdh1、Sdha和Ndufs1)、脂肪酸氧化(如Acamd、Hadh和Echs1)以及成年心脏发育(如Ttn、Myh6和Myh7)相关的蛋白是这一簇的创始成员,表明参与能量代谢和收缩的蛋白质在IRI期间受到乳酸化的严格调控(图1g和补充图2a;补充数据13)。有趣的是,C1还包括与内肽酶和水解酶活性负调控相关的蛋白,包括Serpina3k(SA3K)和Serpina1b(图1g和补充图2a;补充数据13)。第二组蛋白(乳酸化组C2)在MI期间乳酸化增加,随后在再灌注时下降(补充图2a),包括参与ATP和丙酮酸代谢过程的蛋白(如Dld、Gapdh和Pkm),以及参与肌丝滑动和肌节组织的蛋白(如Tnnt2、Ttn和Myh6)(图1g和补充图2a;补充数据13)。第三组(乳酸化组C3)包括肌节蛋白(Csrp3、Ttn、Atp2a2和Myh6)和Ndufs2,其乳酸化变化最小(图1g和补充图2a;补充数据13)。值得注意的是,由于它们在多个位点发生乳酸化,所有三个簇都共享了Ttn和Myh6。乳酸化组中呼吸和收缩组分的过度代表表明,它们是I/R应激期间内源性乳酸化的主要底物。差异表达的蛋白质被分为两个相反的趋势(图1h;补充数据14)。蛋白质组簇1(C1)显示出与乳酸化组C1高度相似的模式,而蛋白质组C2在MI期间略有下降,随后在I/R中迅速下降(补充图2b)。有趣的是,负调控肽酶和水解酶活性的蛋白,包括SA3K、Serpina1b和Serpining1,是上调最强烈的蛋白之一(图1i;补充数据15)。这些蛋白已知作为丝氨酸蛋白酶抑制剂,有助于阻断某些消化酶的活性,并因在血液凝固、炎症和氧化应激中的调控作用而与心血管疾病相关。观察到I/R特异性差异乳酸化和表达的蛋白质均在丝氨酸型内肽酶抑制剂活性中表现出富集,表明丝氨酸蛋白酶家族蛋白可能在I/R中发挥重要作用(补充图2c,补充数据16)。相反,下调的蛋白几乎完全是细胞外基质(ECM)蛋白,表明I/R损伤后ECM发生了巨大的分解和重塑(图1i;补充数据14)。
为了进一步了解I/R中差异乳酸化蛋白之间的关系,作者使用STRING绘制了它们的功能或物理相互作用网络。如预期的那样,存在两组主要的蛋白:一组与肌节结构相关(如Tnnt2、Ttn、Myh7、Myh6和Desmin),另一组与代谢相关(如Ckm、Pfkm、Mdh1、Sdha、Sdhb、Ndufs1、Ndufa8和Slc25a4)(图1j;补充数据17)。两个丝氨酸蛋白酶家族成员,SA3K和Serpina1b,与血液中的蛋白相关,包括Apoa1和Alb。SA3K还与Myh6(心肌α-肌球蛋白重链)和Ckm(肌酸激酶,M型)功能相关,表明SA3K可能在心肌细胞收缩中发挥作用。接下来,作者重叠了MI和I/R中在乳酸化组和蛋白质组中显著变化的蛋白。令人惊讶的是,除了在I/R期间乳酸化状态显著变化的7种蛋白与蛋白质表达显著变化的蛋白之间有重叠外,四组蛋白之间几乎没有重叠,包括血红蛋白α链复合物(Hba)、血红蛋白β成年主要链(Hbb-b1)、白蛋白(Alb)、载脂蛋白A1(Apoa1)、SA3K、Serpina1b和转铁蛋白(Tf)(图1k;补充数据18)。其中,Hba、Hbb-b1、Alb、Apoa1和Tf是血液中高度丰富的蛋白。令人惊讶的是,剩下的两种蛋白是SA3K和Serpina1b,表明它们在I/R中具有突出且特定的作用。SA3是与小鼠Serpina3蛋白家族密切相关的仅有的人类同源物(其中之一是SA3K),已被证明是预测心肌梗死和心力衰竭不良心脏事件的有效指标。因此,作者着手定义SA3K乳酸化在IRI中的病理生理功能及其后果。
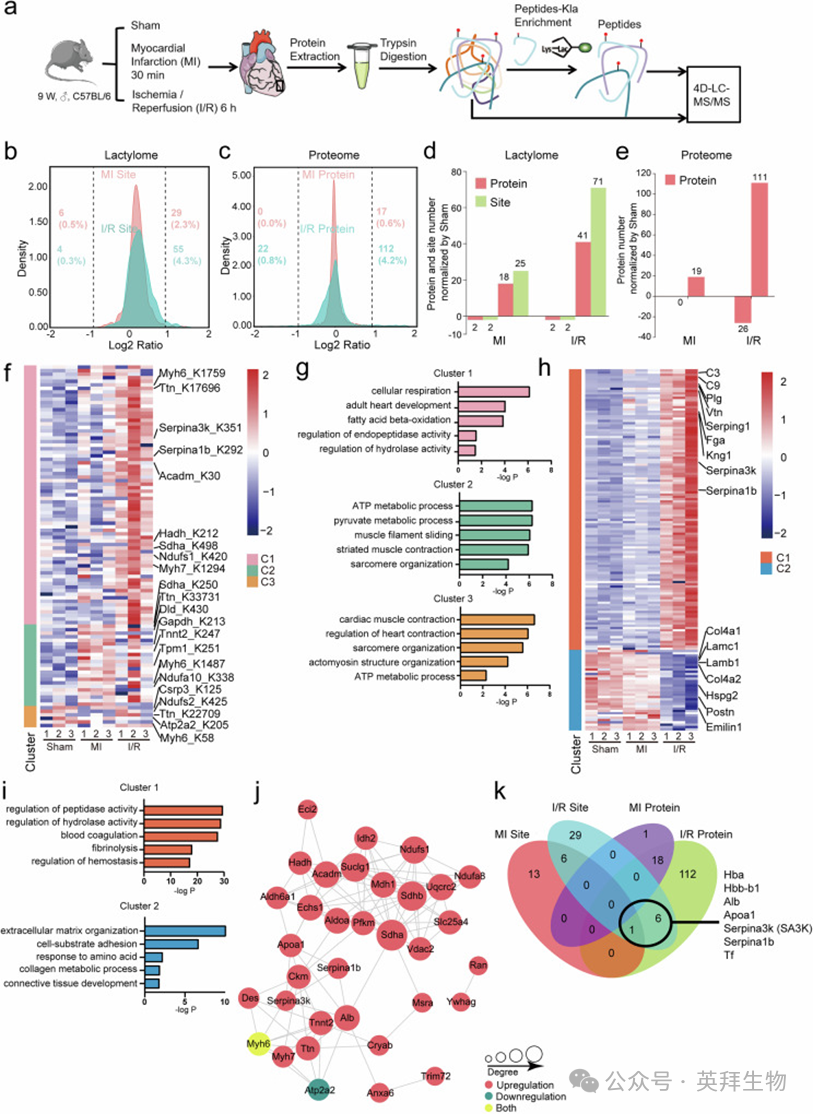
图1. 全局乳酸化组分析心肌缺血/再灌注损伤
2.SA3K及其乳酸化在体内和体外缺血/再灌注损伤中增加
为了验证MI和I/R期间SA3K蛋白表达的变化,作者对MI和I/R小鼠的心肌样本进行了Western blotting分析。与Sham和MI相比,SA3K蛋白水平增加了4.8倍,表明SA3K的增加是IRI特异性的反应(图2a)。一致地,免疫荧光信号强度在I/R小鼠心脏切片中显著高于Sham或MI(图2b)。此外,作者通过免疫沉淀验证了SA3K的赖氨酸乳酸化(SA3K-Kla)。在I/R中,SA3K-Kla显著且特异性增加(图2c,d)。作者的质谱/质谱(MS/MS)数据显示,SA3K的乳酸化发生在残基K351上(补充图3)。为了生成特异性识别SA3K-K351la的多克隆抗体,作者通过点印迹实验验证了抗体的特异性和潜在交叉反应性(补充图4a)。与上述观察一致,使用SA3K-K351la特异性抗体的Western blotting证实了I/R中SA3K-K351la水平的显著增加(图2e)。为了检查SA3K升高的时间过程,作者在I/R 0小时至I/R 6小时之间每小时收集一次心脏组织(补充图4b)。Western blotting显示,与MI相比,K351位点的乳酸化水平在I/R 1小时时显著上升,而蛋白水平在I/R 2小时开始显著增加,并持续至I/R 6小时(补充图4c–e)。这些数据支持在心脏再灌注后,SA3K在K351位点的乳酸化水平迅速且显著升高。
观察到SA3K几乎不与心肌细胞(CM)标记物ACTN2共定位(图2b)促使作者研究增加SA3K表达的细胞基础。免疫荧光显示,在I/R中,FB标记物波形蛋白(VIM)与SA3K有强烈的共定位(图2f)。作者分离了原代新生小鼠CM和CFB,并检查了基础水平的SA3K mRNA表达。与CM相比,SA3K在FB中的表达水平显著更高(图2g)。接下来,作者通过在体外分别培养FB和CM于正常氧(N)、缺氧(H,1%O₂,24小时)或缺氧-再氧(H-N,1%O₂,24小时,随后正常氧12小时)条件下模拟I/R。在FB中,SA3K-K351la和蛋白水平在H-N中特异性且显著上调(图2h)。相比之下,这种增加在CM中不存在(图2i)。为了确定这种现象的保守性,作者分离了成年原代人CM和CFB。与新生小鼠心脏细胞一致,SA3(SA3K的人类同源物)也在CFB中特异性表达(图2j)。其表达也在H-N刺激下在CFB中特异性上调(图2k)。鉴于SA3是一种分泌蛋白,作者进一步通过酶联免疫吸附测定(ELISA)定量培养人CFB的培养液中的SA3,一致地观察到在H-N刺激下分泌的SA3水平显著增加(图2l)。为了进一步探索H-N是否在人CM和CFB中引起SA3-Kla的变化,作者通过免疫沉淀检查了SA3-Kla水平。在人CFB中,SA3-Kla在H-N中显著升高,但在缺氧或CM中则没有(图2m,n)。综上所述,这些发现表明SA3K及其人类同源物SA3在CFB中在蛋白表达和乳酸化水平上增加,这是I/R或H-N损伤的反应。
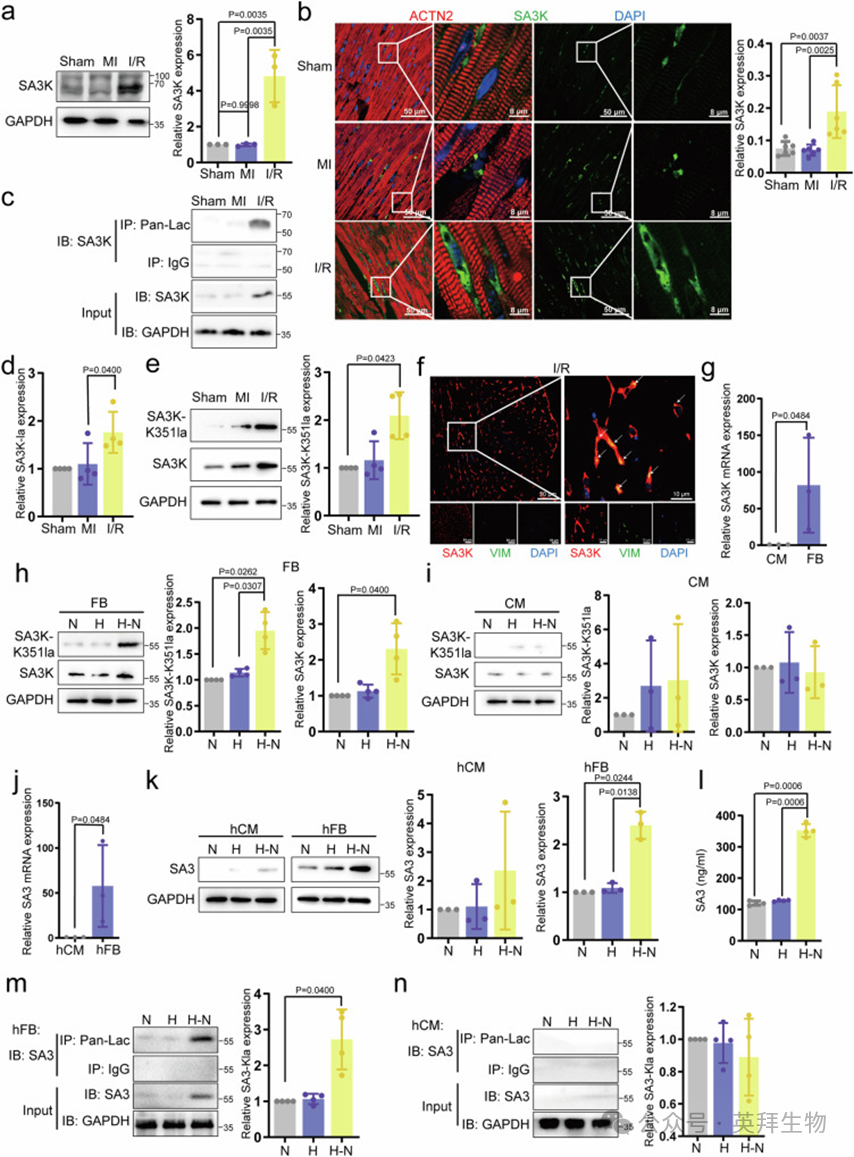
图2. SA3K及其乳酸化在体内和体外缺血/再灌注损伤中增加
3.L-乳酸通过增强乳酸化依赖的蛋白稳定性上调SA3K水平
为了探究SA3K蛋白和乳酸化水平增加的分子基础,作者观察到在小鼠心脏梗死区域的SA3K mRNA水平并未增加(补充图5a)。同样,在体外培养的CM或FB中,H-N处理也未能增强小鼠的SA3K mRNA水平(补充图5b)或人类细胞的SA3 mRNA水平(补充图5c)。这些结果表明,SA3K蛋白水平的升高不太可能是由于转录增加的结果,这促使作者推测其调控可能发生在蛋白水平。
作者还注意到,小鼠心脏梗死区域的全组织L-乳酸显著增加(补充图5d)。同样,体外培养的新生小鼠CM和FB中的细胞内L-乳酸也被H和H-N强烈上调(补充图5e)。这些L-乳酸的变化与乳酸化蛋白和蛋白Kla位点的显著增加相平行(图1f)。鉴于先前的研究报告蛋白Kla水平受乳酸影响,且蛋白修饰广泛参与蛋白翻译后调控,作者假设I/R期间产生的L-乳酸通过K351调节SA3K蛋白的稳定性。
为了验证这一点,作者首先将FB暴露于不同浓度的L-乳酸中,观察到SA3K和SA3K-K351la均呈剂量依赖性增加(图3a),表明L-乳酸诱导了SA3K乳酸化和蛋白表达的增加。由于细胞内L-乳酸的产生依赖于糖酵解和线粒体代谢的平衡,作者测试了这两条通路中的酶活性是否可以调节L-乳酸水平以及随后的SA3K-K351la(图3b)。不出所料,线粒体呼吸链复合体I抑制剂鱼藤酮强烈诱导L-乳酸产生(图3c),而乳酸脱氢酶(LDH)抑制剂草酸显著抑制L-乳酸水平(图3c)。相应地,草酸和鱼藤酮剂量依赖性地降低和增加SA3K-K351la和SA3K水平(图3d,e),进一步暗示乳酸直接调节了SA3K-K351la和SA3K。这些发现表明,外源性和内源性乳酸同时调节了SA3K-K351la和SA3K。SA3K-K351la和SA3K之间的正相关表明,SA3K-K351la促进了SA3K蛋白的稳定性。
为了验证这一观点,作者用cycloheximide(CHX)处理细胞以抑制蛋白合成,并在有无L-乳酸或草酸添加的情况下随时间定量蛋白表达。计算得出的SA3K半衰期从2.98小时增加到6.74小时(L-乳酸处理),而在乳酸被草酸抑制后,半衰期下降到1.92小时(图3f)。在存在蛋白酶体抑制剂MG132和CHX的情况下,蛋白水平保持稳定(图3f),表明乳酸在维持SA3K蛋白稳定性方面发挥了作用。在成人原代人CFB中也观察到类似的现象,L-乳酸处理使SA3的半衰期从4.12小时增加到6.36小时,而草酸处理使SA3的半衰期缩短到2.16小时(补充图5f)。为了进一步评估这种调控是否由SA3K-K351la介导,作者构建了一个突变体,即带有FLAG标签的SA3K(K351R),并将其与野生型(WT)重组蛋白进行了比较。计算得出的外源WT-SA3K的半衰期在L-乳酸处理后从4.66小时增加到6.71小时,而K351R突变体的半衰期几乎与WT蛋白相同(4.84小时)(图3g),表明K351la对于乳酸稳定SA3K是必需的。
为了测试这一观点,作者构建了一个突变体,即带有FLAG标签的SA3K(K351R),并将其与野生型(WT)重组蛋白进行了比较。计算得出的外源WT-SA3K的半衰期在L-乳酸处理后从4.66小时增加到6.71小时,而K351R突变体的半衰期几乎与WT蛋白相同(4.84小时)(图3g),表明K351la对于乳酸稳定SA3K是必需的。
为了排除乙酰化(SA3K-Kac)调节SA3K蛋白稳定性的可能性,作者首先检查了在增加或减少L-乳酸浓度时SA3K-Kac水平的变化。免疫沉淀显示,无论是细胞外L-乳酸(L-乳酸处理)还是细胞内乳酸(草酸或鱼藤酮处理)的变化对SA3K-Kac均无影响(补充图6a)。在培养基中加入100μM乙酰辅酶A略微延长了内源性SA3K的半衰期(3.77小时),但对重组SA3K-WT的半衰期(4.60小时)无影响(补充图6b)。综上所述,这些数据表明L-乳酸通过抑制SA3K的降解来发挥作用,而不是通过乙酰化。
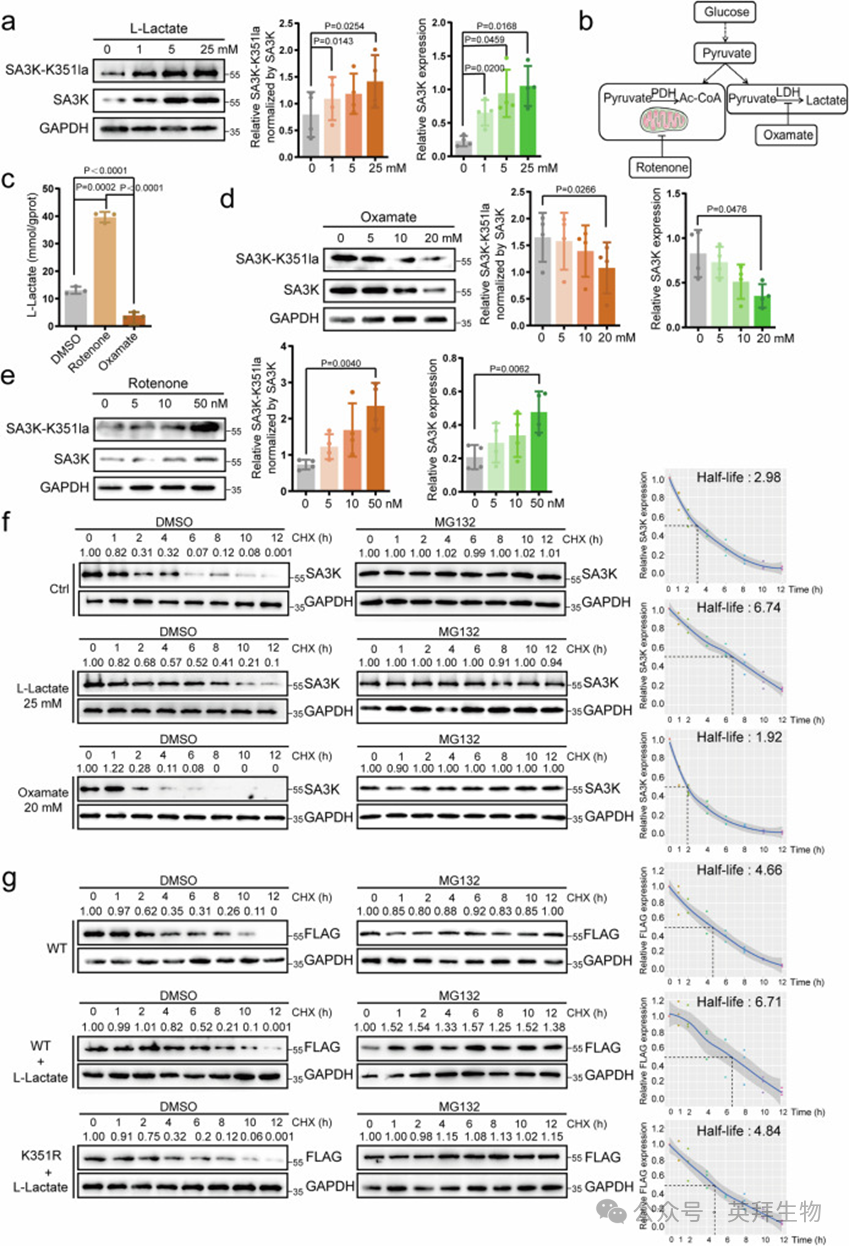
图3. L-乳酸通过增强乳酸化依赖的蛋白稳定性上调SA3K水平
4.SA3K是再灌注损伤中心脏保护的重要驱动因子
为了直接评估SA3K在IRI中的作用,作者生成了SA3K基因敲除(SA3K-KO)小鼠(图4a和补充图7a)。一组野生型(WT)小鼠和两组SA3K-KO小鼠分别被结扎30分钟,然后再灌注6小时以模拟I/R。在第二组SA3K-KO小鼠中,在再灌注时注射了重组纯化的His标签SA3K(0.5 mg/kg)(图4a)。2,3,5-三苯基四氮唑氯化物(TTC)和伊文思蓝染色在I/R后1天显示,各组间风险区域(AAR)相当(图4b,c)。然而,与WT对照组相比,SA3K-KO小鼠的AAR标准化梗死面积显著增加,而通过注射重组SA3K可以挽救(图4d)。为了评估心脏损伤,作者测量了血清中心脏肌钙蛋白T、心脏肌钙蛋白I和肌酸激酶MB(CK-MB)的水平。这些标志物在SA3K-KO小鼠中显著增加,与WT对照组相比,并且通过注射外源性SA3K部分减轻(图4e–g)。通过末端脱氧核苷酸转移酶dUTP缺口末端标记(TUNEL)染色评估的凋亡细胞比例在I/R后1天在SA3K缺陷小鼠中显著增加,而SA3K的注射显著逆转了凋亡的增加(图4h)。在多个时间点(基线、7天、14天和28天)进行超声心动图分析以评估心脏功能变化。SA3K-KO小鼠的基线心脏功能与WT对照小鼠无法区分(补充图7c–h)。在7天、14天和28天时,与WT对照组相比,SA3K缺陷小鼠的射血分数(EF)严重受损,而His-SA3K注射提供了适度的保护效果(图4i,j,补充图7b,c)。类似的趋势在分数缩短(FS)中观察到(图4j,补充图7d)。左心室舒张末期容积(LVEDV)、左心室收缩末期容积(LVESV)、左心室舒张末期内径(LVIDd)或收缩末期内径(LVIDs)在D28时在SA3K-KO小鼠中显著增加,表明I/R后发生心室扩张,而通过补充外源性His-SA3K部分挽救(图4j)。这些参数中的一些在D7时就出现了显著变化(补充图7e–h)。在I/R后28天,作者通过Masson三色染色分析心脏纤维化。与WT心脏相比,SA3K-KO心脏显示出胶原沉积显著增加,而SA3K注射显著减轻了纤维化(图4k和补充图7f)。综上所述,这些发现表明SA3K为心脏提供了针对IRI的保护。
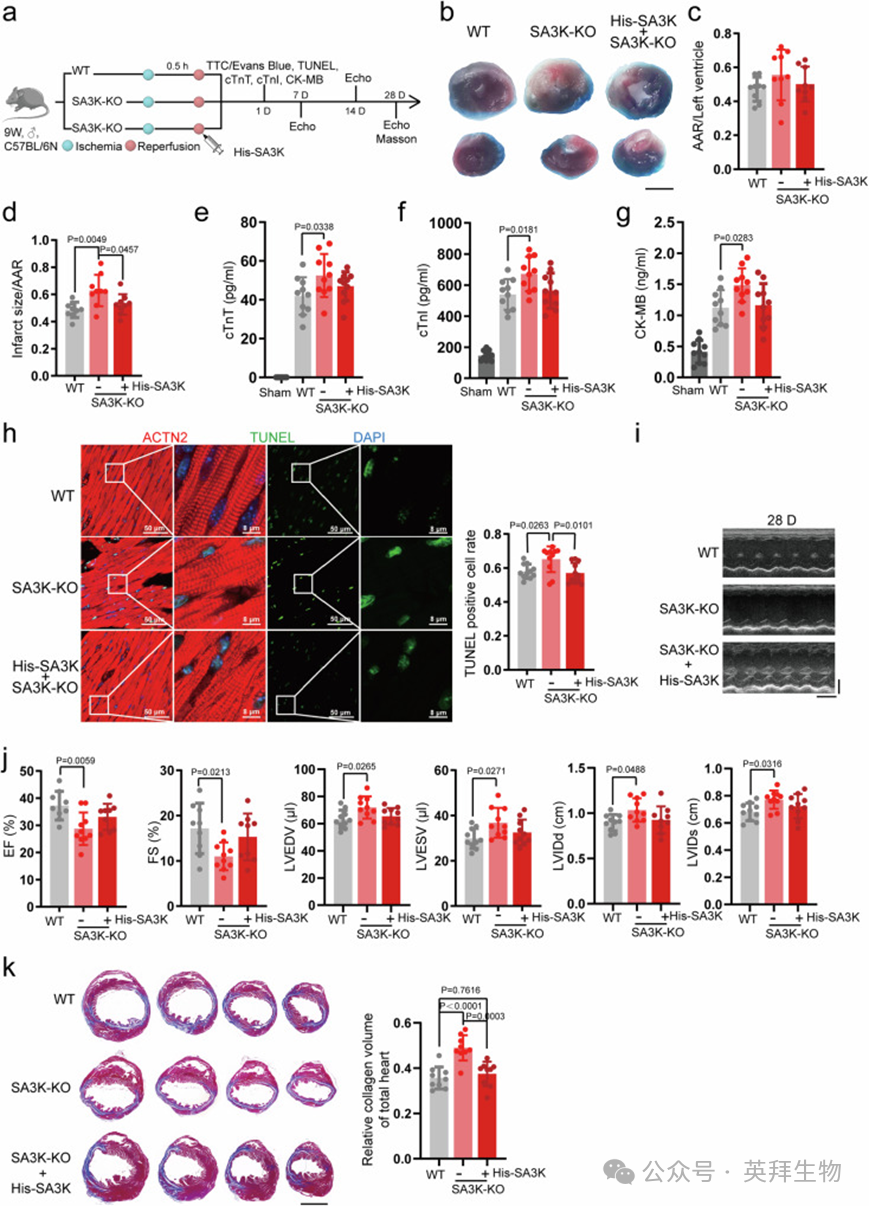
图4. SA3K是再灌注损伤中心脏保护的重要驱动因子
5.SA3K在K351位点的乳酸化对心脏保护至关重要
为了深入了解K351la在IRI中的作用,作者生成了表达野生型SA3K(SA3K-WT)或突变型SA3K(SA3K-K351R)的腺相关病毒血清型9(AAV9)(图5a和补充图8a)。小鼠通过尾静脉注射AAV9。I/R手术4周后,通过TTC染色评估梗死面积。与EV对照组相比,表达WT SA3K的小鼠在I/R后1天的梗死面积显著减少,而表达K351R突变型SA3K的小鼠与EV对照组相比无显著差异,表明K351对于SA3K的保护作用至关重要(图5b–d)。血清中cTnI和CK-MB的水平在表达WT SA3K的小鼠中显著降低,而在表达K351R突变型SA3K的小鼠中则没有(图5e–g)。外源性表达SA3K-WT显著减轻了I/R诱导的凋亡,而SA3K-K351R则未能显示出与EV对照组相比的任何保护作用(图5h)。超声心动图分析显示,病毒注射并未干扰基线心脏功能(补充图8c–h)。SA3K-WT显著改善了心脏功能,这种效果从7天持续到28天,而SA3K-K351R则未能显示出任何改善(图5i,j,补充图8b–h)。最后,28天时的纤维化评估显示,SA3K-WT显著减轻了纤维化,而SA3K-K351R则没有(图5g和补充图8e)。总之,这些数据表明,SA3K在K351位点的乳酸化对于SA3K的心脏保护作用至关重要,表明蛋白半衰期而非蛋白总量决定了IRI的保护。
SA3K通过旁分泌信号保护心肌细胞免受IRI诱导的凋亡。
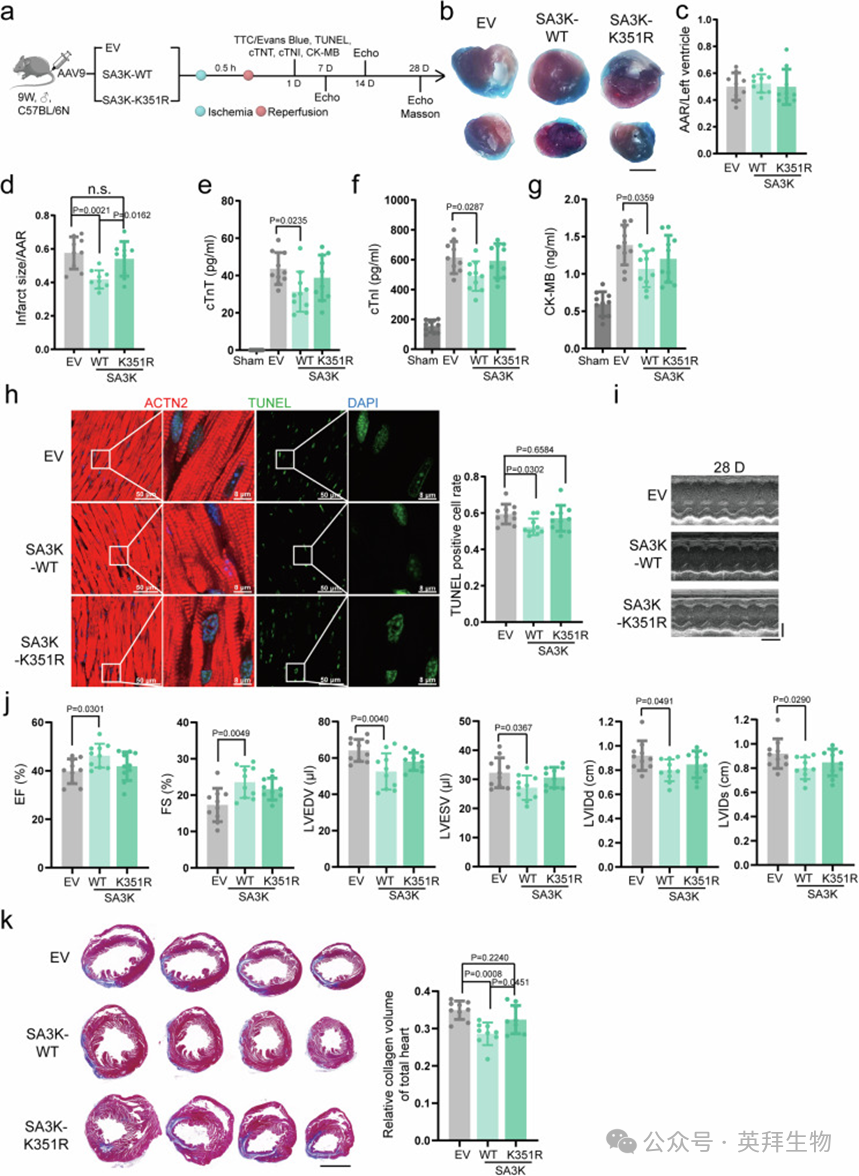
图5. SA3K在K351位点的乳酸化对心脏保护至关重要
6.FBs分泌的SA3K通过旁分泌信号传导保护CM免受IRI诱导的细胞凋亡
重新分析TUNEL染色,特别是针对CM(TUNEL+ACTN2+)的分析揭示了SA3K最显著地改变了CM的凋亡(补充图9a,b)。鉴于CM本身几乎不表达SA3K(图2),并且由于CM特异性的SA3K对H-N的响应最小(图2),作者假设FB通过旁分泌相互作用保护CM免受IRI的影响,主要通过分泌SA3K介导。
为了验证这一假设,作者首先从新生小鼠或成人人类心脏组织中分离出FB和CM,并分别培养(图6a)。将FB在正常氧或H-N条件下培养的条件培养液分别用于CM培养,后者随后接受H-N处理。在一组接受H-N条件培养液的CM中,作者使用SA3K或SA3中和抗体(Nab)来确定SA3K/SA3在旁分泌效应中的作用(图6a)。H-N条件培养液显著抑制了小鼠CM的凋亡,这一效应被Nab抵消(图6b,c)。相应地,小鼠CM中的裂解caspase 3和Bax水平也被H-N条件培养液抑制,而用Nab处理的培养液对CM凋亡的保护作用很小(图6d,e)。在成人原代人FB和CM中也观察到类似的结果(图6d,e)。此外,在新生小鼠和成人原代细胞中,H-N条件培养液显著抑制了乳酸脱氢酶(LDH)的释放,并增强了ATP水平,表明CM的存活率更高,这些效应也被Nab抵消(图6f–h)。然而,细胞活性氧(ROS)和呼吸作用受到的影响最小(补充图9c,补充图10)。这些结果表明,在IRI期间,成纤维细胞(FBs)产生了一种分泌物,可以保护心肌细胞(CMs)免受IRI诱导的凋亡,这种效应主要通过分泌的SA3K/SA3介导。
为了进一步研究SA3K在IRI中的保护作用,作者通过慢病毒介导的敲低在FBs中耗尽SA3K,并将条件培养液转移到CM培养液中,随后对CM进行H-N处理(图6i和补充图9d,e)。与非靶向(NT)对照组相比,shSA3K#1显著增加了细胞群中凋亡细胞的比例,而shSA3K#2也显示出增加的趋势(图6j,k)。同样,SA3K的耗尽还增强了裂解caspase 3和Bax的表达,以及LDH的释放(图6l,m,补充图9f)。然而,ATP水平、ROS和线粒体功能并未受到影响(补充图9g,h,补充图10)。在成人原代人CFBs中耗尽SA3也产生了类似的结果(图6l,n,补充图9d–h)。因此,这些发现表明,SA3K/SA3由FBs分泌,能够保护CMs免受IRI诱导的凋亡。
此外,作者通过慢病毒介导的表达或向CM培养液中添加重组His-SA3K,直接在CMs中增加SA3K的表达(补充图11a)。在H-N刺激后,观察到CMs受到显著保护,表现为凋亡标志物下调(补充图11b,c)。这一发现表明,SA3K能够保护CMs免受IRI诱导的凋亡。
为了了解这种保护是否依赖于K351位点的乳酸化,作者在FBs中异位表达SA3K-WT和SA3K-K351R,并评估它们的保护效率(图6o,补充图9i,j)。与体内数据类似,SA3K-WT显著抑制了CMs的凋亡,而SA3K-K351R未能显示出任何效果(图6p,q)。SA3K-K351R也未能抑制裂解caspase 3和Bax的表达,或LDH的释放,而WT形式则可以(图6r,补充图9k)。再次,ATP水平、ROS和线粒体呼吸并未受到影响(补充图9l,m),表明SA3K对细胞死亡的调节并非通过线粒体损伤介导。综上所述,这些结果强调了Kla在CMs中的调节作用。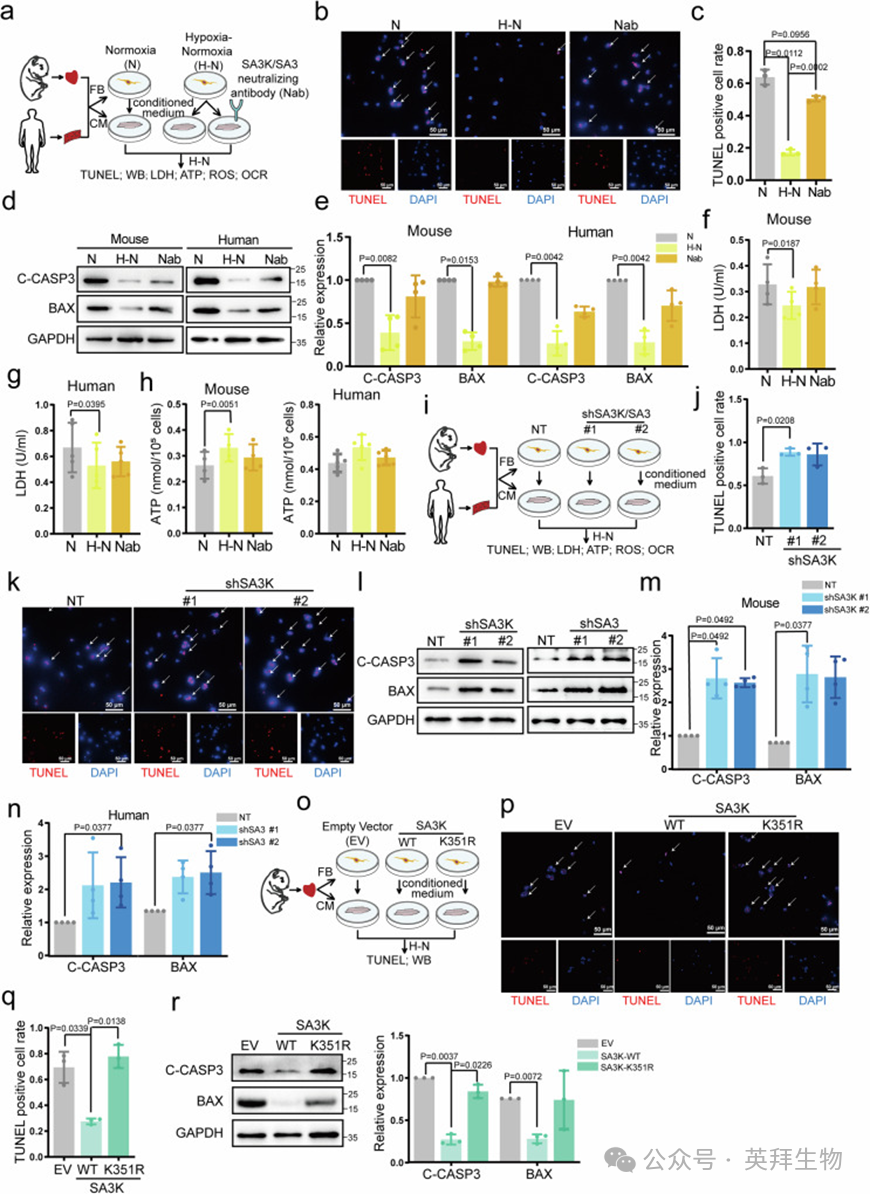
图6. FBs分泌的SA3K/SA3通过旁分泌信号传导保护CM免受IRI诱导的细胞凋亡
7.SA3K通过抑制WNT通路并激活RISK和SAFE通路保护CMs
为了探索SA3K保护作用的分子机制,作者利用上述实验设计(图6i,o),并分析了CMs中改变的信号通路。作者首先分析了AMPK信号通路,这是一条经典的保护性心肌通路。然而,SA3K/SA3水平未能调节小鼠或人类CMs中AMPK信号通路的活性(补充图12a–f)。进一步地,鉴于SA3K已被报道在血管生成中抑制WNT信号通路,作者试图确定WNT通路是否在SA3K水平变化后发生改变。在小鼠FBs中过表达SA3K或在成人原代人FBs中过表达SA3,可导致CMs中pLRP和β-catenin水平显著下降(补充图12g–i)。相反,当使用条件培养液处理时,来自SA3K耗尽的FBs的培养液使pLRP显著增加,而β-catenin也显示出增加的趋势(补充图12g–i)。使用成人原代人CFBs的条件培养液处理成人原代人CMs时,观察到类似但较弱的效果(补充图12g–i)。
鉴于SA3K的促存活效应,作者进一步测试了它是否促进经典的保护性RISK和SAFE通路。RISK通路是一组促存活激酶通路,包括AKT和ERK1/2,它们赋予心脏对IRI的保护作用。在小鼠FBs中过表达SA3K或在成人原代人FBs中过表达SA3,导致相应物种CMs中p-AKT和p-ERK信号显著上调(图7a–d)。相反,SA3K的沉默显著抑制了小鼠CMs中AKT和ERK1/2的磷酸化。在人类细胞中观察到类似的趋势,但只有shSA3#1的p-ERK达到了统计学意义(图7c,d)。SAFE通路涉及JAK和STAT-3的激活,已被证明在小鼠、猪和人类中IRI保护中发挥关键作用。在小鼠FBs中过表达SA3K显著增加了N-H刺激的CMs中JAK2和STAT3的磷酸化(图7e,f)。相反,SA3K的耗尽导致了相反的趋势。在成人原代人细胞中也观察到类似的结果,表现为在shSA3#2耗尽时STAT3磷酸化显著抑制(图7g,h)。综上所述,这些数据表明,SA3K通过抑制WNT信号通路并激活RISK和SAFE保护性通路来保护CMs免受IRI诱导的凋亡。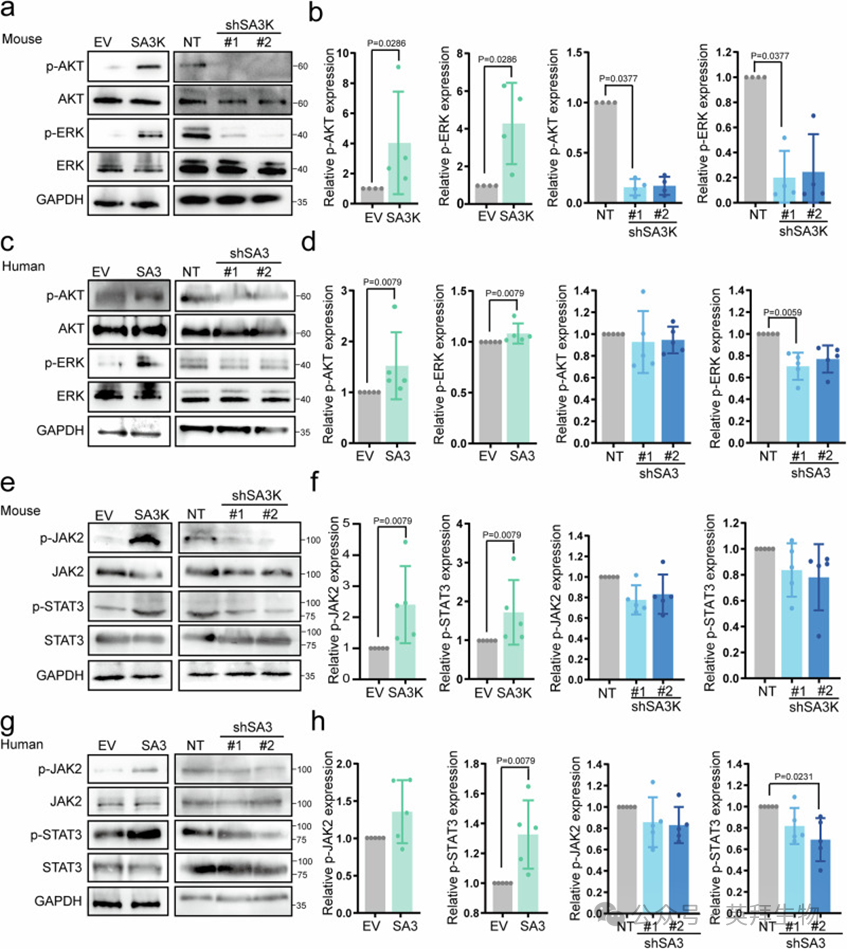
图7. SA3K通过抑制WNT通路并激活RISK和SAFE通路保护CMs
参考文献
1.Wang L, Li D, Yao F, Feng S, Tong C, Rao R, Zhong M, Wang X, Feng W, Hu Z, Jin B, Wang L, Hu S, Zhou B. Serpina3k lactylation protects from cardiac ischemia reperfusion injury. Nat Commun. 2025 Jan 25;16(1):1012.