






G9a-TRIM21轴介导的 FoxO3a 降解促进氧化应激和焦亡,加重糖尿病肾缺血再灌注损伤
糖尿病性肾缺血再灌注损伤(RIRI)是一种严重的手术并发症,在糖尿病患者中预后尤其差。组蛋白甲基转移酶G9a与多种病理过程有关,但其在糖尿病RIRI中的作用尚不清楚。新出现的证据表明,非组蛋白甲基化可能在缺血性损伤的细胞反应中起关键作用。我们发现G9a在糖尿病RIRI模型中表达显著上调。在体内和体外模型中,G9a基因缺失减轻了肾损伤,减少了氧化应激和焦亡。在机制上,G9a直接与FoxO3a在赖氨酸262位点相互作用并使其甲基化,从而促进其被E3泛素连接酶TRIM21识别。TRIM21随后介导了赖氨酸176位点FoxO3a的k48连锁多泛素化,导致蛋白酶体降解。G9a介导的FoxO3a降解促进氧化应激、NLRP3炎性体激活和焦亡。重要的是,BIX-01294对G9a的药理学抑制或FoxO3a过表达可显著改善糖尿病RIRI。我们的研究揭示了糖尿病RIRI中一个新的G9a-TRIM21-FoxO3a调节轴,其中G9a介导的甲基化允许FoxO3a泛素化和降解,从而促进焦亡和氧化应激。这些发现表明G9a是预防或治疗糖尿病肾缺血再灌注损伤的潜在治疗靶点。本文于2025年12月发表于Redox Biology(IF=11.9)上。
技术路线:

结果:
1)DM加重RIRI,上调G9a表达
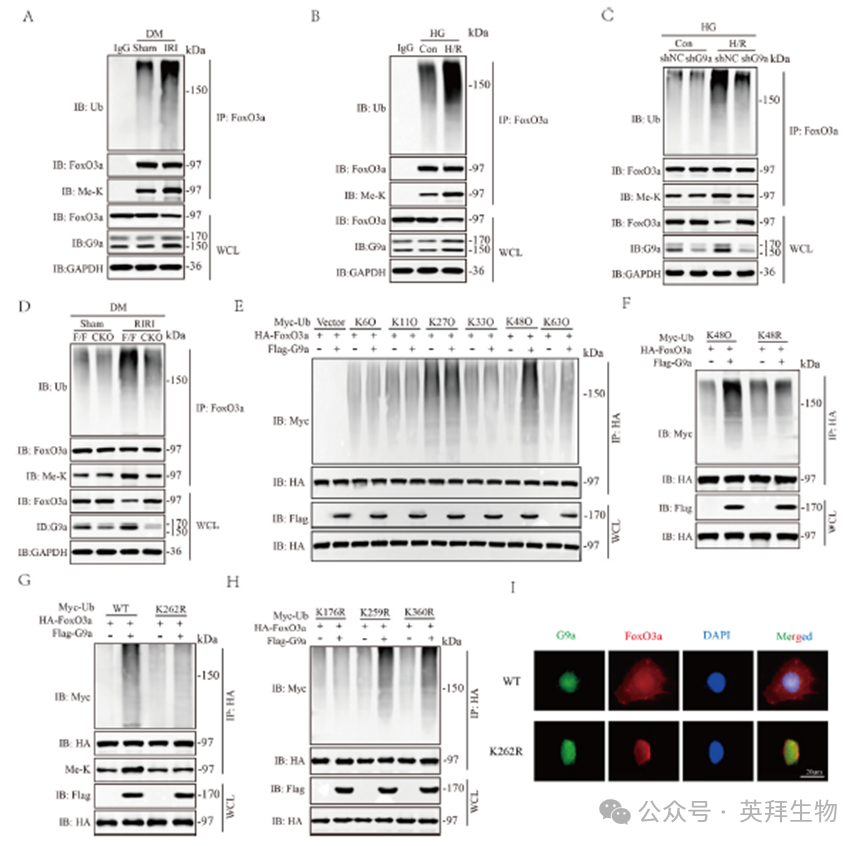
首先,我们建立正常和糖尿病状态下的RIRI动物模型,确认DM对RIRI的易感作用。如图1A-D所示,RIRI后小鼠肾小管扩张,上皮细胞脱离,肾小管结构紊乱,消失,肾损伤评分显著升高,血清BUN和Cr水平显著升高。在DM条件下建立RIRI模型时,上述病理损伤表现和肾功能损害进一步加重,说明DM小鼠更容易发生RIRI。WB结果显示,RIRI后肾损伤标志物KIM1和NAGL蛋白表达增加,DM条件下进一步显著增加(图1E)。体外细胞H/R损伤实验显示,HG培养的HK2细胞系中,KIM1和NAGL蛋白的表达也有类似的增加趋势(图1F)。TUNNEL染色阳性率升高证实HG条件下细胞H/R模型诱导细胞死亡,MDA含量升高,SOD活性降低(图1G)。非组蛋白甲基化在糖尿病RIRI中尚不清楚,因此我们通过RNA测序来观察甲基转移酶的总体表达。RNA测序结果显示,许多蛋白甲基转移酶的表达水平发生了显著变化(图1H)。KEGG通路分析和GSEA富集分析显示,差异表达基因主要富集于炎症、细胞死亡、局灶黏附等相关通路(图1I和J)。基于前人的研究,我们选择G9a来证实其在糖尿病RIRI中的表达。WB实验显示HG作用下DM小鼠和HK-2中G9a的上调均增强,这可能是由转录因子Klf4介导的,直接结合并转激活G9a启动子(图1K和L)。
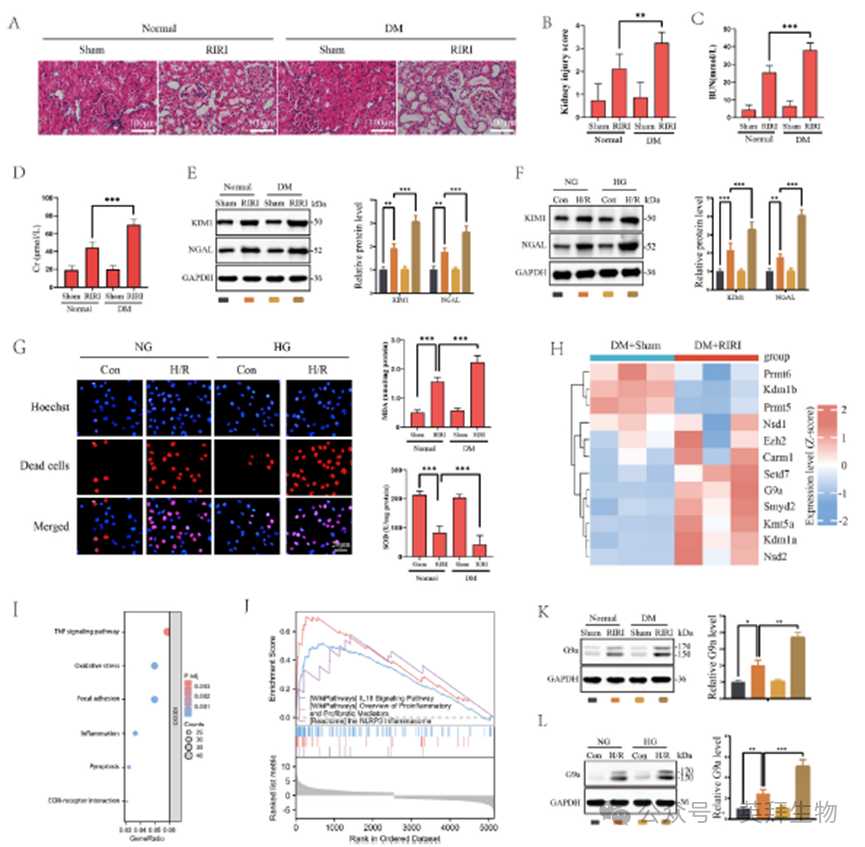
2)抑制G9a可减轻糖尿病RIRI和氧化应激
RNA测序发现,一些具有非组蛋白甲基转移酶活性的甲基转移酶基因的表达水平在糖尿病RIRI中出现了显著改变(图1H)。其中,G9a引起了我们的兴趣。在我们之前的研究中,我们已经证明G9a通过抑制Sirt1来促进RIRI。然而,其在DM相关RIRI中的功能尚不确定。然后检测G9a在体内和体外模型中的表达水平。两种IRI模型的G9a水平均升高,高糖条件下G9a水平进一步升高(图1K和L)。为了阐明G9a在DM相关RIRI中的调控作用,我们在肾小管上皮细胞中产生G9a的条件敲除(CKO)小鼠,通过PCR验证小鼠基因型,并通过WB实验证实敲除效果(图2A、B、C和D)。与G9aFlox/Flox小鼠相比,CKO小鼠在IRI治疗后表现出较低的病理损伤,并伴有肾损伤评分(图2E和F)、血清BUN和Cr水平(图2G和H)以及KIM1和NAGL蛋白表达水平的降低,表明敲除G9a显著减轻了RIRI(图2I)。结果表明,用BIX沉默G9a或抑制G9a活性可导致KIM1和NAGL蛋白表达降低,细胞死亡率降低,MDA含量降低,SOD活性升高,表明氧化应激减轻,肾小管上皮细胞损伤减轻(图2J-M)。
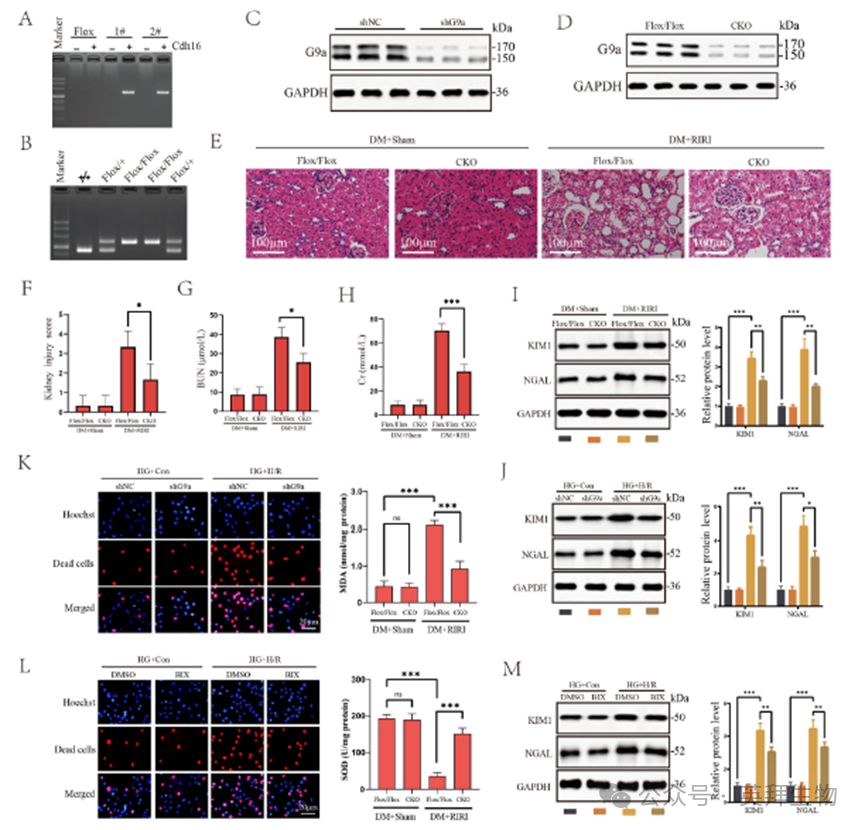
3)G9a在糖尿病RIRI中促进细胞焦亡并靶向FoxO3a
为了进一步阐明G9a在糖尿病RIRI中的潜在调控机制,我们进行了质谱(MS)实验和Co-IP实验以鉴定与G9a相互作用的蛋白,并对这些互作蛋白进行了KEGG富集分析。通过MS实验,我们鉴定出FoxO3a是G9a作用的下游靶点,且KEGG分析显示细胞焦亡通路在所有互作蛋白中显著富集(图3A和B)。我们使用Necrostatin-1、ABT-199和VX-765评估了细胞活力的变化,发现经焦亡抑制剂处理后,KIM1和NAGL蛋白显著降低,而细胞活力显著提高。同时,在体内和体外模型中,观察发现焦亡相关标志物NLRP3、ASC、Cleaved-caspase-1和IL-1β的表达增加,且在DM条件下其表达进一步被促进(图3C)。此外,在RIRI过程中,与G9aFlox/Flox小鼠相比,CKO小鼠的NLRP3、ASC、Cleaved-caspase-1和IL-1β蛋白表达显著下调(图3D)。沉默G9a或使用BIX抑制G9a活性,可导致体外焦亡相关蛋白显著下调(图3E)。随后,在293T细胞中过表达G9a和FoxO3a进行的Co-IP实验以及GST pull-down实验,证实了G9a与FoxO3a之间存在直接相互作用(图3F–H)。此外,通过表达G9a和FoxO3a的截短突变体,我们确定了G9a与FoxO3a之间的相互作用结构域。Co-IP结果显示,G9a蛋白的680-896氨基酸(aa)区段与FoxO3a蛋白的150-300aa区段相互作用(图3I和J)。
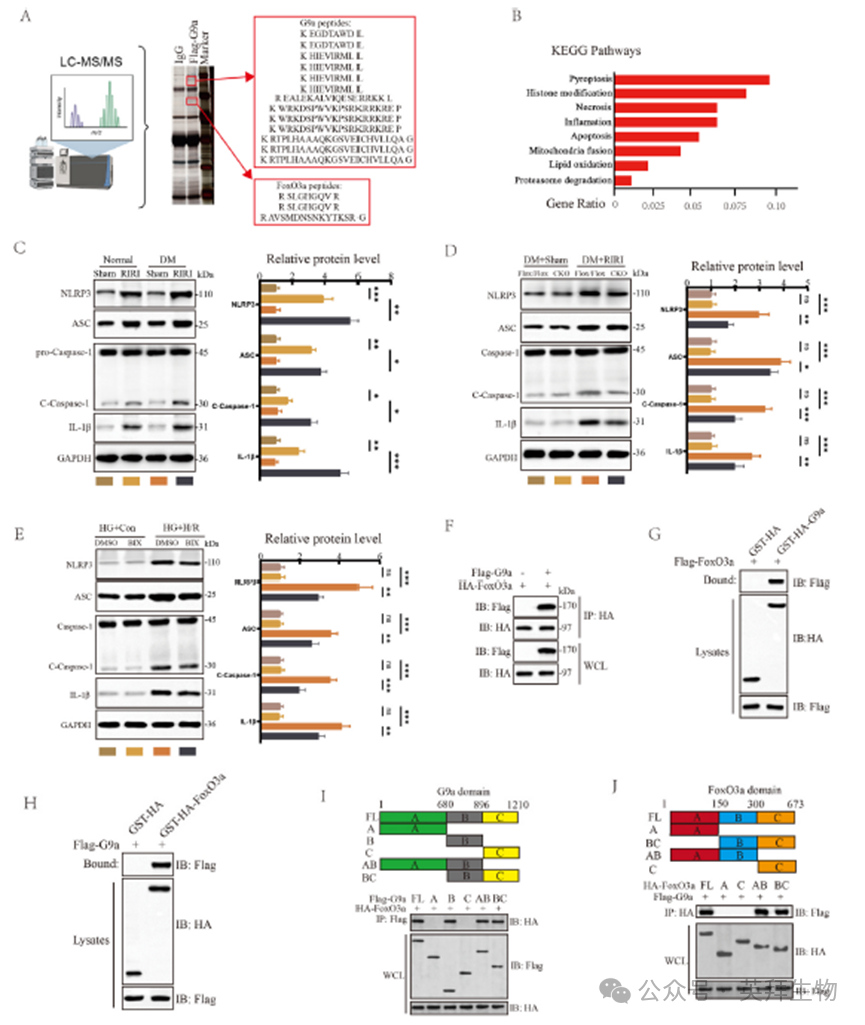
4)G9a通过甲基化促进FoxO3a的降解
在确定了G9a和FoxO3a之间的直接相互作用后,我们研究了G9a在DM相关RIRI中对FoxO3a表达的调节作用。在细胞H/R模型的HG条件下,FoxO3a水平下降更为明显(图4A)。沉默G9a或用BIX抑制G9a活性可导致FoxO3a蛋白表达显著上调(图4B和C)。通过体外过表达系统,我们发现G9a可以直接甲基化FoxO3a(图4E)。此外,我们试图通过位点定向诱变确定G9a对FoxO3a的特异性甲基化位点。结果表明K262是G9a甲基化FoxO3a的位点(图4F)。我们进一步研究了G9a如何调节FoxO3a的稳定性。结果表明,CQ不能维持FoxO3a蛋白的稳定性,而MG132能显著稳定FoxO3a蛋白(图4J)。时间过程分析进一步表明,G9a降低了内源性FoxO3a的半衰期(图4K)。这些结果表明G9a通过蛋白酶体-泛素系统调节FoxO3a蛋白的表达。我们还检测了G9a对HK-2细胞中FoxO3a mRNA的影响,结果显示shG9a和BIX处理均未影响FoxO3a的转录(图4G-I)。综上所述,这些发现表明G9a通过在K262位点直接甲基化FoxO3a与DM相关的RIRI中的FoxO3a相互作用,从而促进FoxO3a的蛋白酶系统降解。
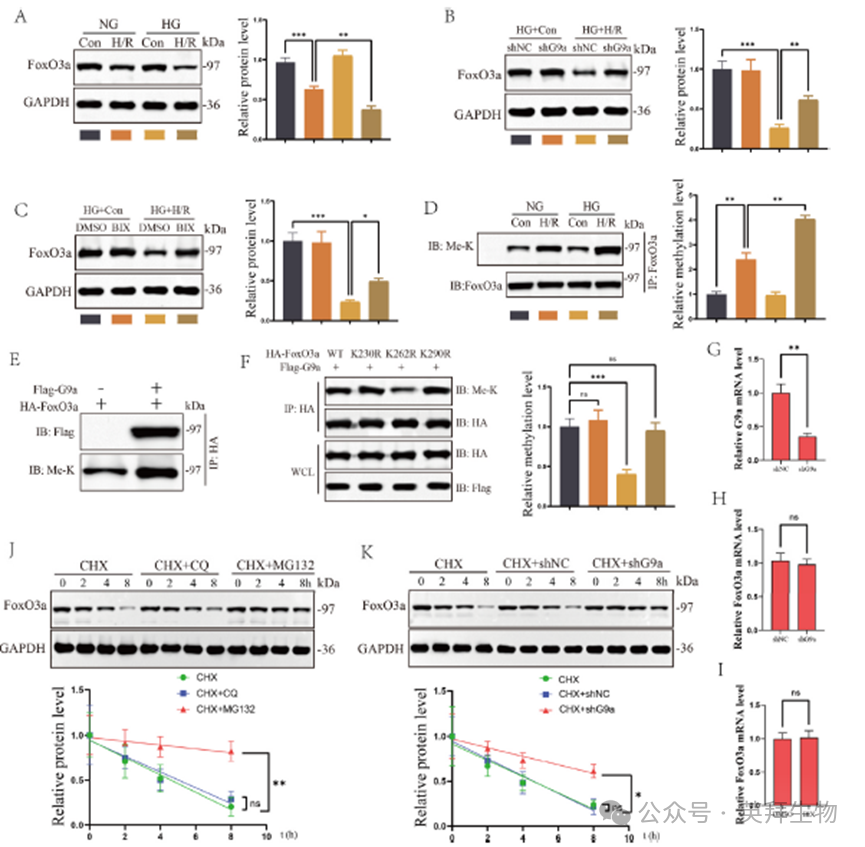
5)G9a促进k48连接的FoxO3a在K176位点的泛素化和蛋白酶体降解
进一步的研究表明,FoxO3a的泛素化在体内和体外的糖尿病RIRI中增加(图5A和B),而G9a的缺失或沉默降低了FoxO3a的泛素化(图5C和D)。由于不同类型的多泛素连接介导不同的生物学功能,我们在HK-2细胞中表达了HA标记的FoxO3a和各种泛素(k60、K11O、K27O、K33O、K48O和K63O),每种泛素只含有一个特定的赖氨酸残基,可用于多泛素链的形成。泛素化结果显示,G9a通过形成k48链增强FoxO3a的泛素化降解(图5E)。当K48泛素化位点发生突变时,G9a介导的FoxO3a泛素化被消除(图5F)。此外,将FoxO3a的甲基化修饰位点K262突变并转染到293T细胞后,我们发现,在甲基化失活后,FoxO3a的泛素化被显著抑制(图5G)。随后,通过生物信息学分析,我们确定了FoxO3a蛋白上三个潜在的泛素化位点,并对三个位点分别进行突变后检测FoxO3a的泛素化位点。结果表明,K176是FoxO3a的泛素化修饰位点(图5H)。通过免疫荧光双染色观察G9a和FoxO3a的亚细胞定位。结果显示,在WT-G9a的激活下,FoxO3a在糖尿病RIRI中表现出易位的倾向,而这种倾向被K262R-G9a消除(图5I)。总的来说,这些发现证明G9a在糖尿病RIRI期间调节FoxO3a蛋白甲基化,从而促进FoxO3a的泛素化和易位。
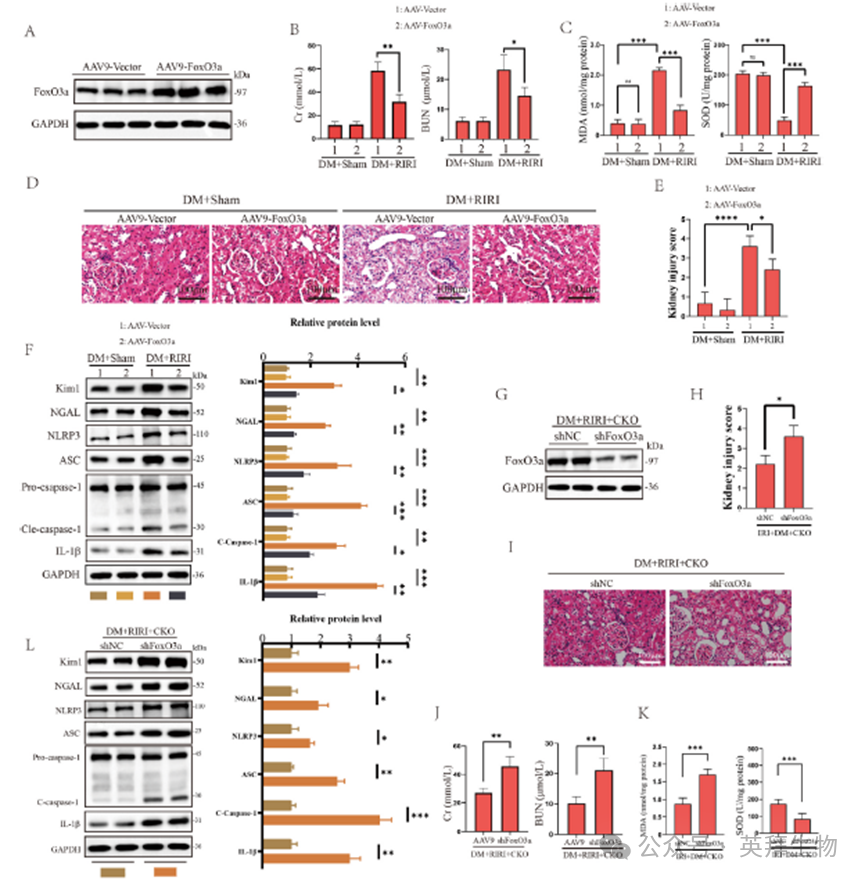
6)FoxO3a减轻糖尿病RIRI,并参与G9a驱动糖尿病RIRI
接下来,我们利用AAV9在体内过表达FoxO3a,研究FoxO3a在糖尿病RIRI中的关键作用(图6A)。结果显示,DM + IRI组FoxO3a过表达导致血清BUN、Cr、MDA水平显著降低,SOD含量升高,减轻肾小管上皮细胞的组织病理学损伤,降低肾损伤标志物KIM1和NAGL的表达,从而显著减轻RIRI的组织损伤和肾功能障碍(图6B-E)。WB分析显示,在RIRI期间,FoxO3a的过表达显著抑制了NLRP3、ASC、Cle-caspase-1和IL-1β蛋白的表达(图6F)。为了阐明G9a是否通过FoxO3a调控DM相关的RIRI,我们使用AAV9在G9a敲除小鼠中沉默FoxO3a。我们的研究结果显示,沉默FoxO3a导致血清Cr和BUN显著升高,小管扩张和上皮细胞脱落,进一步加剧肾损伤(图6G-K)。此外,我们观察到沉默FoxO3a显著增加了肾损伤标志物KIM1和NAGL的表达,以及焦亡标志物NLRP3、ASC、Cle-caspase-1和IL-1β的表达(图6L)。同时,FoxO3a的沉默导致Cr、BUN和MDA水平升高,SOD含量降低(图6J和K)。总之,这些结果为G9a-FoxO3a轴在糖尿病RIRI进展过程中促进细胞焦亡和氧化应激的潜在参与提供了证据。
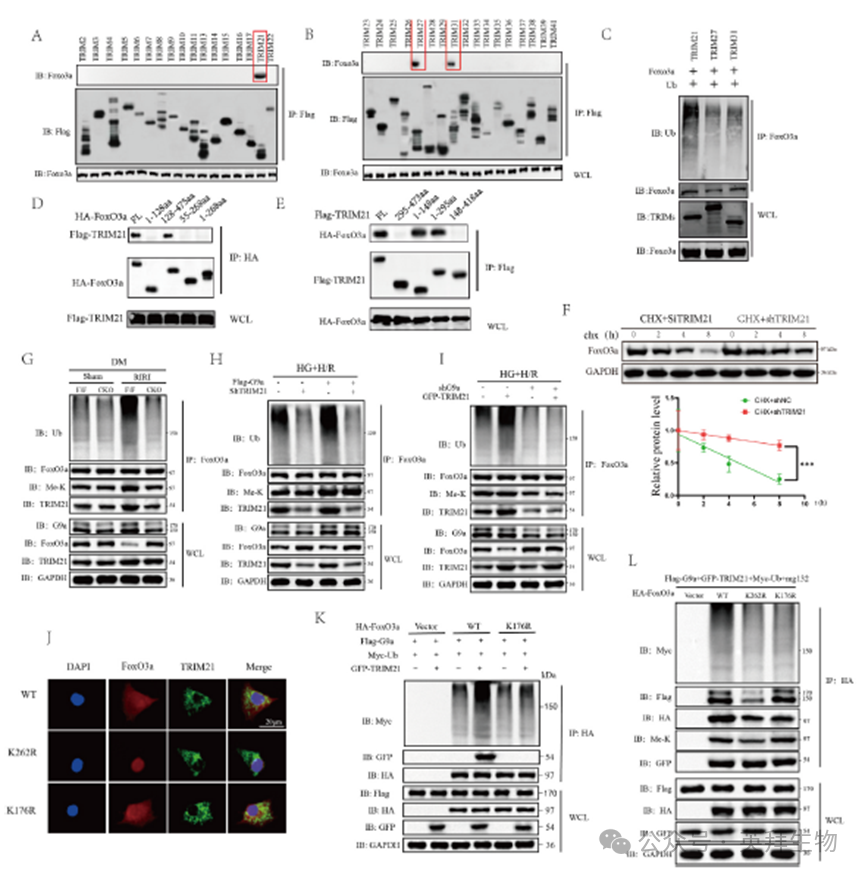
7)G9a促进TRIM21介导的FoxO3a泛素化
为了进一步阐明G9a的作用机制,我们对多个TRIM蛋白进行了IP筛选,以鉴定与FoxO3a相关的E3泛素连接酶。在初步筛选中,TRIM21、TRIM27和TRIM231在IP中显示出与FoxO3a连接的条带(图7A、B)。因此,选择这三个TRIM蛋白进行Co-IP泛素化分析。结果表明,只有TRIM21增强了FoxO3a的泛素化(图7C),泛素化位点为K176(图7K)。环己亚胺CHX实验表明,TRIM21的抑制有效延长了FoxO3a的半衰期(图7F)。为了阐明G9a甲基化与TRIM21介导的泛素化之间的关系,我们进行了一系列综合实验。我们的研究结果表明,缺血条件显著增强FoxO3a的泛素化,这可以通过有条件地敲除G9a来消除(图7G)。此外,细胞实验表明,TRIM21敲低可抑制H/R条件下FoxO3a的泛素化,而FoxO3a的甲基化状态保持不变(图7H)。相反,在H/R条件下,TRIM21的过表达增强了FoxO3a的泛素化。然而,G9a敲除导致的FoxO3a甲基化水平的降低同时阻碍了TRIM21介导的FoxO3a泛素化(图7I)。K262突变诱导的甲基化抑制消除了泛素化。相反,K176R突变抑制泛素化而不影响FoxO3a的甲基化状态(图7L)。免疫荧光分析显示,K262R能够限制FoxO3a的核转位,而K176R不阻碍FoxO3a与TRIM21的共定位(图7J)。总之,这些发现表明TRIM21促进FoxO3a的泛素化,其活性通过G9a介导的甲基化调节。
结论:
我们的研究首次发现,G9a 以非组蛋白形式甲基化 FoxO3a,导致其发生位移,并通过 TRIM21 介导的 K48 连接泛素化,最终导致 FoxO3a 的降解。这一过程在氧化应激和糖尿病RIRI的预后中发挥着调节作用。这些效应与 FoxO3a 上的甲基化位点 K262 和泛素化位点 K176 密切相关。开发 G9a 抑制剂或 FoxO3a 激动剂对于治疗糖尿病 RIRI 具有重要意义。因此,我们的研究结果为糖尿病 RIRI 的治疗提供了关键且新颖的策略。
参考文献:
Zheng Q, Qin X, Huang S, Yan Z, Liu J, Xiong Y, Zhao X, Ni X, Mei H, Jian J, Wang J, Lu Q, Chen Z, Liu X, Wan S, Liu H, Wang L. The G9a-TRIM21 axis exacerbates diabetic renal ischemia-reperfusion injury by inducing methylation-dependent ubiquitination and degradation of FoxO3a to promote oxidative stress and pyroptosis. Redox Biol. 2025 Dec 8;89:103964. doi: 10.1016/j.redox.2025.103964.