






YTHDC2通过铜死亡抑制肺癌对EGFR-TKI的抵抗
尽管第三代EGFR-TKIs如奥希替尼显著提高了非小细胞肺癌(NSCLC)患者的生存率,但获得性耐药仍是主要的临床挑战,其潜在机制尚不完全清楚。在本研究中,我们发现与奥希替尼敏感的患者来源的异种移植(PDX)组织和肺癌细胞系相比,奥希替尼耐药的组织和细胞系中 YTHDC2 的表达显著下调。进一步的研究表明,YTHDC2 通过促进铜死亡来克服肺癌细胞对奥希替尼的耐药性。从机制上讲,YTHDC2 以 m6A 依赖的方式与铜转运蛋白 SLC31A1 的 mRNA 中的 m6A 修饰位点结合。这种相互作用增强了 SLC31A1 mRNA 的稳定性和蛋白质表达,从而增加了肿瘤细胞内的铜转运并诱导铜死亡。此外,我们发现铜离子载体DSF通过增加 YTHDC2 的表达来克服奥希替尼耐药性。总的来说,我们的研究结果阐明了YTHDC2-SLC31A1-铜死亡轴是EGFR-TKI耐药的关键机制,并提出了逆转该耐药性的新治疗策略。本文于2025年12月发表于Oncogene(IF=7.3)上。
技术路线:

结果:
1)YTHDC2是EGFR-TKI奥西替尼耐药的关键调节因子
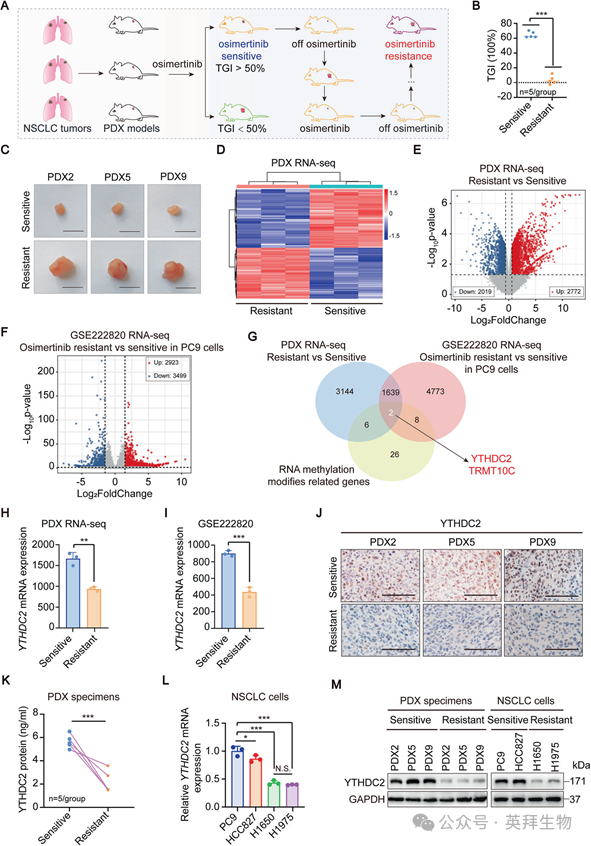
为探究EGFR-TKI奥希替尼耐药性的分子机制,我们首先利用人非小细胞肺癌(NSCLC)组织建立了奥希替尼耐药和奥希替尼敏感的患者来源异种移植(PDX)模型。模型构建的示意图见图1A。我们生成了对奥希替尼获得性耐药的PDX模型。基于TGI指数,我们选择了五个敏感和五个耐药的PDX模型进行进一步研究(图1B),其代表性图像见图1C。接下来,我们对来自奥希替尼敏感和耐药PDX小鼠的肿瘤组织进行了RNA测序(RNA-seq)。分析了两组间的差异表达基因(DEGs)(图1D, E)。考虑到RNA修饰在奥希替尼耐药中的作用尚不完全清楚,我们将PDX RNA-seq的DEGs与公共数据集GSE222820(图1F)中的基因以及已知的RNA甲基化相关基因进行了交集分析。值得注意的是,只有YTHDC2和TRMT10C被确定为在所有三个来源中都重叠的基因(图1G),这表明它们可能与奥希替尼耐药性相关。为了进一步研究奥希替尼耐药性的潜在调控因子,我们分析了PDX来源的RNA-seq数据和公共数据集GSE222820中YTHDC2和TRMT10C的表达水平。与其各自的敏感对照组相比,YTHDC2的表达在奥希替尼耐药的PDX组织和PC9耐药细胞中均显著下调(图1H, I)。鉴于YTHDC2在数据集中的一致性及其与耐药性的负相关,我们优先选择YTHDC2进行机制研究。为验证这些发现,我们采用多种正交方法来评估YTHDC2的表达。免疫组化(IHC)染色(图1J)、ELISA定量(图1K)、RT-qPCR分析(图1L)和Western blot(WB)检测(图1M)共同证明,与敏感对照组相比,奥希替尼耐药PDX组织中的YTHDC2水平显著降低。这些结果为YTHDC2作为EGFR-TKI治疗中奥希替尼耐药性的关键调控因子提供了有力证据。
2)YTHDC2抑制奥西替尼耐药性
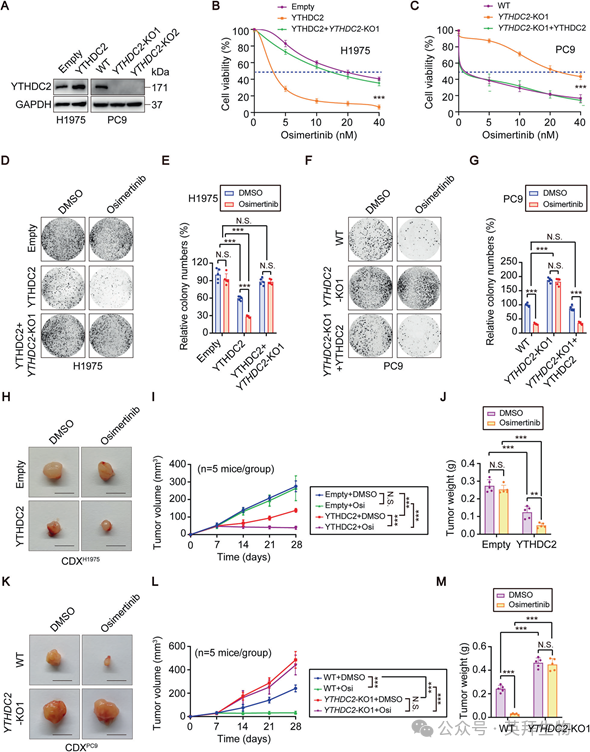
接下来,我们研究了YTHDC2在奥西替尼耐药中的功能作用。首先,我们生成了YTHDC2稳定过表达的H1975细胞和YTHDC2敲除的PC9细胞,证实了这些操作的效率(图2A)。在奥西替尼治疗后,我们使用CCK-8测定法评估细胞活力。在H1975细胞中,与对照组相比,YTHDC2过表达显著降低了细胞活力,增加了奥西替尼的敏感性(图2B)。然而,当YTHDC2在这些过表达细胞中被敲除时,奥西替尼敏感性降低,恢复耐药性(图2B)。相反,在PC9细胞中,YTHDC2敲低显著增强细胞活力,并赋予更高的奥西替尼耐受性(图2C)。值得注意的是,在这些敲除细胞中重新引入YTHDC2表达逆转了获得性耐药,恢复了奥西替尼的敏感性(图2C)。菌落形成分析进一步证实了这些发现。在H1975细胞中,经奥西替尼处理后,YTHDC2过表达组形成的菌落明显少于对照组;然而,在这些过表达细胞中敲除YTHDC2可恢复集落形成(图2D,E)。相比之下,与对照组相比,敲除YTHDC2的PC9细胞在集落数量上没有显著差异,但重新引入YTHDC2显著减少了集落的形成(图2F,G)。总之,这些结果表明,YTHDC2过表达逆转了奥西替尼耐药性,而其缺失促进了体外耐药性。接下来,我们在体内评估了这些效应。CDXH1975肿瘤的代表性图像(图2H)所示,经奥西替尼治疗后,YTHDC2过表达组的肿瘤体积(图2I)和重量(图2J)均较对照组显著减小。而对于CDXPC9肿瘤(图2K),经奥西替尼治疗后,YTHDC2敲除组的肿瘤体积(图2L)和重量(图2M)与对照组无显著差异。这些发现表明,YTHDC2过表达在体内可逆转奥西替尼耐药,而敲除YTHDC2可促进体内耐药。
3)YTHDC2通过铜死亡调节奥希替尼耐药性
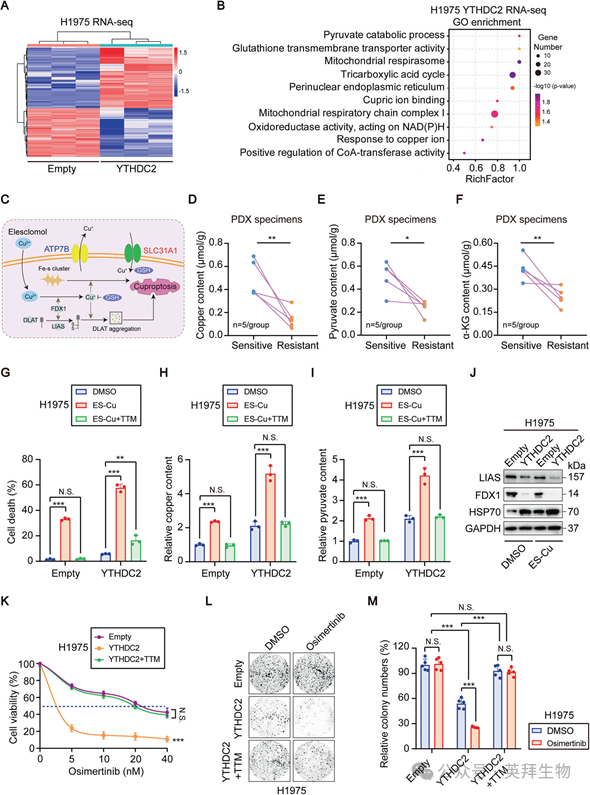
我们的研究结果表明,YTHDC2在EGFR-TKI奥希替尼耐药性中发挥着关键的调控作用。为了阐明其潜在的分子机制,我们对过表达YTHDC2的H1975细胞和携带空载体的对照细胞进行了RNA测序。热图(图3A)描绘了差异表达基因。随后的GO功能分析(图3B)显示,在“铜离子结合”和“铜离子反应”等生物学过程中存在显著富集。这些过程都与铜死亡相关,表明YTHDC2对奥希替尼耐药性的调控可能涉及铜死亡。图3C提供了说明铜死亡分子机制的示意图。为了确立YTHDC2通过调控铜死亡来影响奥希替尼耐药性的作用,我们首先研究了其调节铜死亡相关代谢通路的能力。我们定量分析了奥希替尼耐药和敏感PDX组织中的关键铜死亡相关代谢物——铜、丙酮酸和α-酮戊二酸(α-KG)。如图3D–F所示:与敏感组织相比,耐药PDX组织中的铜水平显著降低(图3D),丙酮酸含量明显更低(图3E),α-KG水平也有所下降(图3F)。这些发现表明,奥希替尼耐药性与铜死亡相关代谢活性的抑制有关。接下来,我们检测了在使用铜死亡诱导剂elesclomol-CuCl2(ES-Cu)或铜螯合剂四硫钼酸盐(TTM)处理后,YTHDC2对细胞死亡和代谢物水平的影响。在过表达YTHDC2的细胞中,与对照组相比,ES-Cu处理显著增加了细胞死亡(图3G)、细胞内铜水平(图3H)、丙酮酸含量(图3I)。这些增加在很大程度上被TTM的共处理所消除。此外,在ES-Cu处理后,与对照组相比,YTHDC2过表达的细胞中铜死亡生物标志物LIAS和FDX1的表达显著降低,而HSP70的表达则显著增加(图3J)。最后,为了确立YTHDC2介导的铜死亡与奥希替尼耐药性之间的因果关系,我们测试了阻断铜死亡是否能逆转YTHDC2过表达所带来的增敏效应。在奥希替尼处理后,向YTHDC2过表达的细胞中添加TTM显著提高了细胞活力(图3K)和集落形成能力(图3L, M)。这些发现证实,YTHDC2通过调控铜死亡来调节奥希替尼的耐药性。
4)铜转运体SLC31A1是YTHDC2的靶标
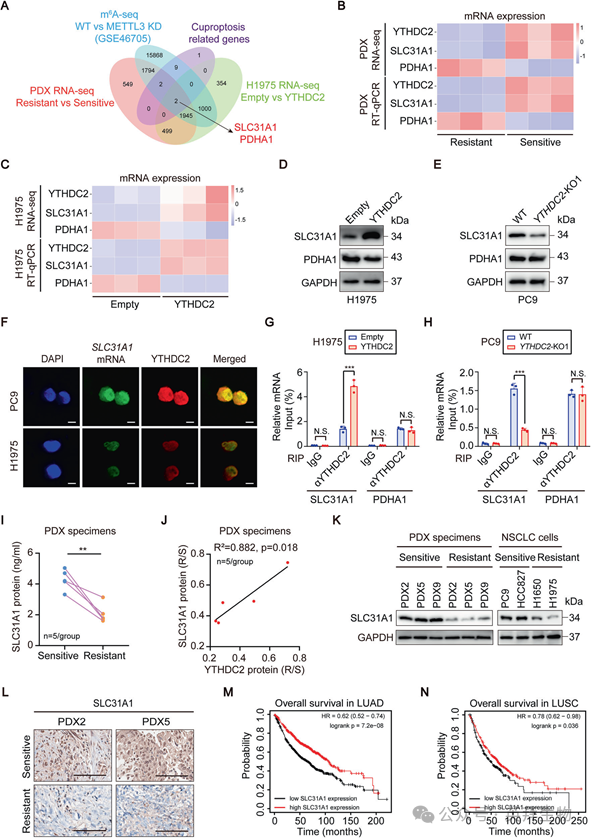
接下来,我们研究了YTHDC2通过铜死亡调控奥希替尼耐药的分子机制。作为m6A修饰的“阅读器”,YTHDC2需要结合m6A修饰的mRNA才能发挥的功能。因此,我们试图确定YTHDC2目标。为此,我们综合分析了METTL3敲低(KD, GSE46705)后的m6A-seq数据、PDX RNA-seq数据、H1975 RNA-seq数据和铜裂相关基因。将这些数据集相交后,只有SLC31A1和PDHA1是共有的(图4A)。为了进一步阐明YTHDC2与其可能的靶点SLC31A1和PDHA1之间的关系,我们利用PDX RNA-seq和RT-qPCR分析了YTHDC2、SLC31A1和PDHA1在PDX组织中的mRNA表达水平。与敏感对照相比,在奥西替尼耐药的PDX组织中,SLC31A1表达显著降低,而PDHA1表达升高(图4B)。值得注意的是,YTHDC2过表达上调了SLC31A1 mRNA水平,但PDHA1 mRNA保持不变(图4C)。为了在蛋白水平上验证这些发现,我们评估了YTHDC2过表达或敲除后SLC31A1和PDHA1的表达。YTHDC2过表达显著增加了SLC31A1蛋白水平,而PDHA1蛋白丰度没有检测到变化(图4D)。相比之下,YTHDC2敲除降低了SLC31A1蛋白的表达,而PDHA1蛋白水平保持稳定(图4E)。
为了研究YTHDC2与其可能的靶点SLC31A1之间的相互作用,我们首先在PC9和H1975细胞中使用RNA FISH检测了它们的细胞内定位。RNA FISH显示,在PC9细胞中,YTHDC2和SLC31A1 mRNA的共定位更强,而在H1975细胞中,这种相互作用明显较弱(图4F)。为了证实直接结合,我们进行了RIP测定。结果表明,YTHDC2特异性结合SLC31A1 mRNA,但不与PDHA1 mRNA相互作用(图4G,H)。这些发现确立了SLC31A1是YTHDC2的直接靶点。接下来,ELISA分析显示,与敏感对照相比,奥西替尼耐药PDX组织中SLC31A1蛋白表达显著降低(图4I)。此外,SLC31A1蛋白水平与YTHDC2蛋白表达呈正相关(图4J), WB(图4K)和IHC(图4L)验证了这种关系。Kaplan-Meier生存曲线显示,SLC31A1低表达患者的生存期明显长于LUAD(图4M)和LUSC(图4N)高表达患者。总之,这些发现确定SLC31A1是YTHDC2的直接靶点,其下调有助于奥西替尼耐药,并可作为肺癌的独立预后标志物。
5)YTHDC2通过靶向SLC31A1调控铜死亡
在确定了YTHDC2通过铜死亡调控奥西替尼耐药性以及SLC31A1是YTHDC2的靶点后,我们接下来研究了YTHDC2是否通过SLC31A1调控铜死亡。SLC31A1过表达或敲除后,H1975和PC9细胞中铜死亡标志物FDX1和HSP70的WB分析显示,与对照组相比,SLC31A1过表达后,FDX1显著降低,HSP70显著升高。相反,SLC31A1敲除后观察到相反的效果(图5A)。细胞内铜(图5B)和丙酮酸(图5C)水平的测量显示,SLC31A1过表达时升高,SLC31A1敲除时降低(图5D,E)。在体内,将SLC31A1敲除的PC9细胞或野生型(WT)细胞皮下注射到胸腺裸小鼠体内产生CDXPC9肿瘤。SLC31A1基因敲除组与对照组相比,肿瘤体积(图5F、G)、肿瘤重量(图5H)、铜含量(图5I)均显著降低。接下来,通过拯救实验证明SLC31A1介导YTHDC2对铜死亡的调控。在DMSO或ES-Cu处理后,对YTHDC2-过表达细胞、SLC31A1敲除的YTHDC2过表达细胞或空载体对照中细胞死亡(图5J)、铜(图5K)和丙酮酸(图5L)水平的测量显示,SLC31A1敲除在很大程度上逆转了YTHDC2过表达的影响。最后,从YTHDC2敲除的PC9细胞、SLC31A1过表达的YTHDC2敲除的细胞或WT PC9细胞中产生CDXPC9肿瘤。代表性肿瘤图像(图5M)、肿瘤体积(图5N)和铜离子水平(图5O)显示,SLC31A1过表达挽救了ES-Cu治疗后YTHDC2基因敲除的效果。这些数据共同证明YTHDC2通过靶向SLC31A1调控铜死亡。
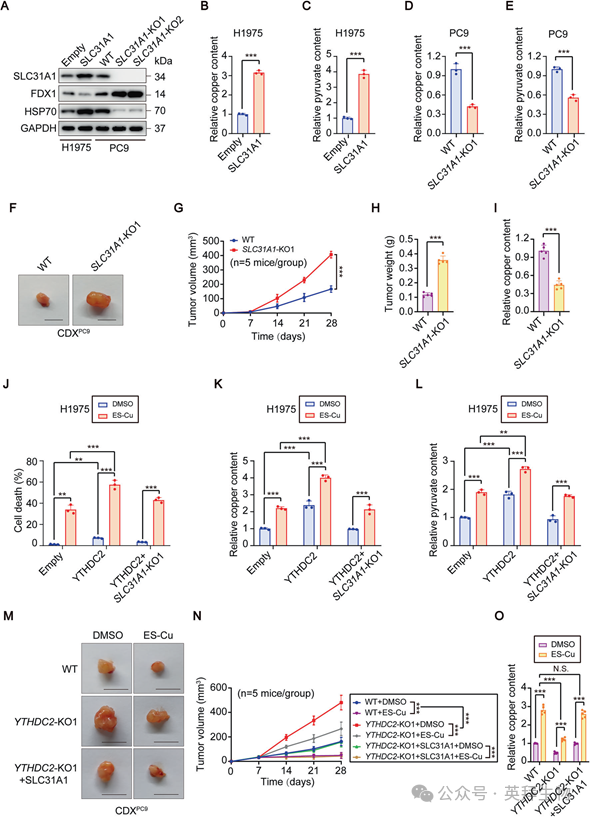
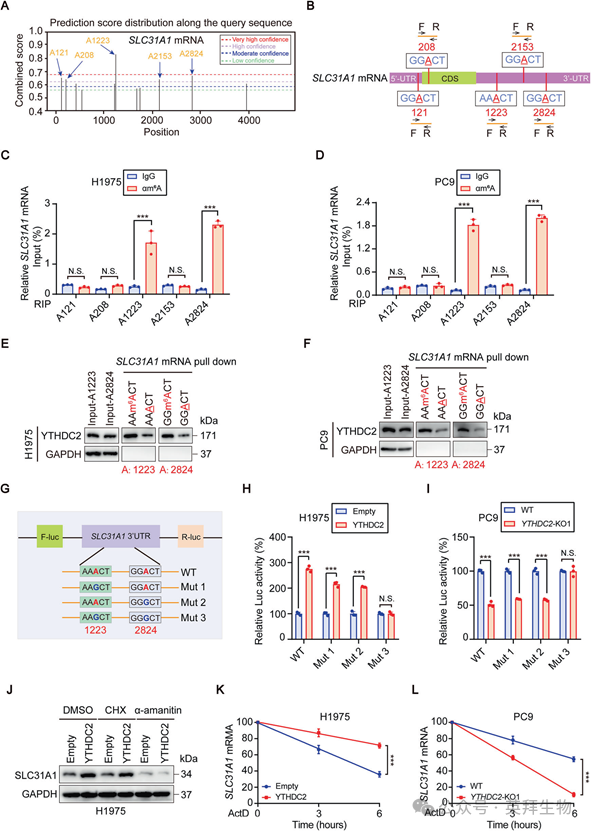
6)YTHDC2结合m6A修饰的SLC31A1 mRNA,维持SLC31A1 mRNA的稳定性
作为m6A读取器,YTHDC2需要与m6A修饰的mRNA结合。因此,我们在YTHDC2结合的SLC31A1 mRNA中鉴定了m6A位点。使用SRAMP在线数据库进行预测,在位置A121、A208、A1223、A2153和A2824上发现了高置信度和非常高置信度的m6A基序(图6A,B)。对H1975和PC9细胞系的MeRIP-qPCR分析显示,m6A仅在含有A1223和A2824的区域显著富集(图6C,D)。使用生物素化的m6A一致性探针(在A1223或A2824处有或没有m6A甲基化)进行RNA下拉试验证实,YTHDC2特异性结合SLC31A1 mRNA中的这些位点(图6E,F)。为了验证这些结合位点,我们构建了SLC31A1 3 ' -UTR中预测的m6A位点的突变体(A-to-G)(图6G)。荧光素酶报告基因检测显示,这些位点的突变显著降低了H1975细胞中YTHDC2过表达诱导的相对荧光素酶活性(图6H),并消除了PC9细胞中YTHDC2敲除的作用(图6I),证实了A1223和A2824是与YTHDC2结合的功能性m6A位点。由于YTHDC2主要调控靶mRNA的翻译效率或稳定性,我们研究了其对SLC31A1的影响。过表达YTHDC2或对照的H1975细胞(图6J)中,经DMSO、环己亚胺(CHX,蛋白质合成抑制剂)或α-amanitin (RNA聚合酶II抑制剂)处理后,SLC31A1蛋白的WB分析显示,SLC31A1蛋白水平仅在α-amanitin处理后才发生变化。这表明YTHDC2可能影响SLC31A1 mRNA的稳定性。为了验证这一假设,我们使用RT-qPCR测量了过表达YTHDC2的H1975细胞或对照组(图6K),以及YTHDC2敲除或WT的PC9细胞(图6L),在Actinomycin D(转录抑制剂ActD)处理0、3和6小时后的SLC31A1 mRNA衰减率。与各自的对照组相比,过表达YTHDC2的细胞(图6K)和敲除YTHDC2的细胞(图6L)中SLC31A1 mRNA衰减速度明显较慢。这些结果表明YTHDC2与m6A修饰的SLC31A1 mRNA结合并增强其稳定性。
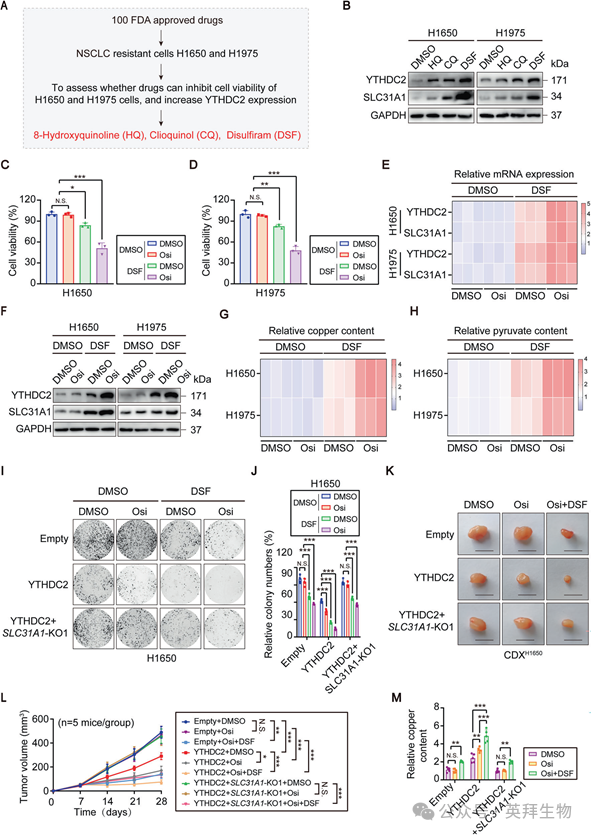
7)DSF通过靶向YTHDC2调节铜死亡,改善奥西替尼耐药性
在确定YTHDC2通过m6A依赖性结合SLC31A1 mRNA调节铜死亡介导奥希替尼耐药后,我们筛选了FDA批准的靶向YTHDC2的小分子药物来克服耐药。筛选了100个化合物的文库,以确定它们抑制H1650和H1975细胞活力和增加YTHDC2表达的能力,鉴定出8-Hydroxyquinoline(HQ)、Chloroquine(CQ)和双硫醚(DSF)是最有效的抑制剂(图7A)。对HQ、CQ和DSF处理的H1650和H1975细胞中YTHDC2和SLC31A1蛋白及mRNA水平的分析显示,三种药物均能提高表达,其中DSF的效果最好(图7B)。因此,我们选择DSF进行进一步的研究。然后,细胞活力测定表明,与DSF共处理显著增强了H1650(图7C)和H1975(图7D)细胞对奥希替尼的敏感性。在用DMSO或DSF治疗奥希替尼后,与对照组相比,DSF处理组中YTHDC2和SLC31A1的mRNA(图7E)和蛋白(图7F)水平,以及细胞内铜(图7G)和丙酮酸(图7H)水平均上调。最后,拯救实验进一步证实了DSF的作用机制。代表性菌落图像(图7I)和定量分析(图7J)表明,DSF通过靶向YTHDC2克服了耐药性。同样,与单独使用奥西替尼相比,DSF和奥西替尼治疗来自YTHDC2过表达细胞的CDXH1650肿瘤(图7K中的代表性图像)导致肿瘤体积(图7L)和铜含量(图7M)减少。然而,在SLC31A1敲除YTHDC2过表达细胞衍生的肿瘤中,这种DSF介导的增敏作用显著减弱。综上所述,这些发现表明DSF靶向YTHDC2调节铜死亡并克服奥西替尼耐药性。
结论:
我们的研究阐明了对EGFR-TKIs获得性耐药的新机制。我们发现YTHDC2以m6A依赖的方式靶向SLC31A1,通过促进铜死亡克服肺癌细胞中的奥希替尼耐药。这些发现为管理对EGFR-TKIs的获得性耐药提供了新的见解和潜在的治疗策略。
参考文献:
Luo J, Xu X, Chen Y, Huang Y, Huang Y, Zhang Y, Ma L, Chen T. YTHDC2 inhibits the resistance of lung cancer to EGFR-TKI through cuproptosis. Oncogene. 2025 Dec 16. doi: 10.1038/s41388-025-03660-1. Epub ahead of print. PMID: 41402633.