






m6A修饰与DNA修复“跨界”联动:胶质母细胞瘤TMZ耐药的新机制被揭示
胶质母细胞瘤(GBM)是一种侵袭性强、易复发且预后不良的恶性肿瘤。尽管替莫唑胺(TMZ)是GBM治疗的基石,但其疗效常因固有或获得性耐药而受限,这凸显了揭示分子机制、发现新的治疗靶点以及开发创新治疗策略的紧迫性。在本研究中,我们发现TMZ耐药性GBM细胞核内丝状肌动蛋白(F-actin)的形成增加。我们还证明,TMZ耐药性GBM细胞中FSCN1的过表达会促进F-actin的形成,并加速DNA双链断裂(DSBs)的修复。进一步研究揭示,TMZ耐药性GBM细胞中YTHDC1的表达显著下降,而该蛋白正是通过m6A修饰调控FSCN1的。此外,FSCN1通过招募FGD1激活CDC42GTP,从而激活CDC42/N-WASP/Arp2/3信号通路,进而驱动核内F-actin的形成。重要的是,将FSCN1抑制剂NP-G2-044与TMZ治疗联合使用,在体外和体内均产生了更强的抗肿瘤效应。综上所述,该研究证实GBM中的核F-actin形成促进了DSB修复,并揭示了通过NP-G2-044靶向FSCN1可能是提高治疗效果、改善GBM患者预后的有前景的策略。该研究于2026年3月发表在《Advanced Science》,IF:14.1。
技术路线

主要研究结果:
1.核F - Actin增加促进GBM细胞对TMZ的耐药性
肿瘤细胞肌动蛋白细胞骨架的变化会影响其行为和生物功能。为了探究TMZ耐药性胶质母细胞瘤(GBM)细胞核内肌动蛋白的改变,我们使用Lifeact和一种带有核定位信号(NLS)的GFP标记质粒(NLS)的GFP标记质粒(Lifeact-EGFP-2xNLS)对GBM细胞系(LN229、U251、HG7)及其相应的TMZ耐药GBM细胞系(229R、251R、HG7R)进行了免疫荧光染色。三维视图结果显示,与亲代GBM细胞相比,TMZ耐药GBM细胞中的核F-actin含量增加,且Lifeact-EGFP-2xNLS染色的特异性优于鬼笔环肽染色(图1A;图S1A)。我们通过免疫荧光染色定量测定了细胞核内F-actin阳性细胞的比例。结果显示,TMZ耐药性GBM细胞中核内F-actin阳性细胞的比例显著增加(图S1B)。随后,吡啶-肌动蛋白实验表明,TMZ耐药性GBM细胞的核提取物比亲本GBM细胞的核提取物更能促进F-actin聚合(图1B;图S1C、D)。F-actin/G-肌动蛋白测定显示,TMZ耐药性GBM细胞核提取物中的F/G肌动蛋白比值也高于亲本GBM细胞(图1C;图S1E)。为验证这些发现,我们利用Lifeact-EGFP-2xNLS对内源性核肌动蛋白进行可视化,并通过共聚焦显微镜的Airyscan模式对其进行成像。我们观察到,在TMZ耐药性GBM细胞中,核F-actin显著增加(图1D)。
接下来,我们旨在探讨核内F-actin在TMZ耐药性中的作用。为抑制核内肌动蛋白聚合,将TMZ耐药性GBM细胞预先用1 µM拉特鲁库林B(LatB)处理30分钟。与二甲基亚砜(DMSO)处理组相比,该处理显著减少了TMZ耐药性GBM细胞中核内F-actin的形成(图1E–G;图S1F–I)。此外,为探究F-actin与TMZ耐药性的关联,我们在抑制F-actin形成后对细胞进行TMZ处理,随后进行流式细胞术和集落形成实验。结果显示,LatB对核内肌动蛋白聚合的抑制对TMZ耐药性GBM细胞的凋亡或增殖无显著影响。然而,与仅接受TMZ处理的细胞相比,同时接受TMZ和LatB处理的TMZ耐药性GBM细胞显示出更高的凋亡率和更低的集落形成能力(图1H、I;图S1J、K)。这些结果表明,核F-actin的形成和功能在GBM细胞TMZ耐药性的发展中起着关键作用。
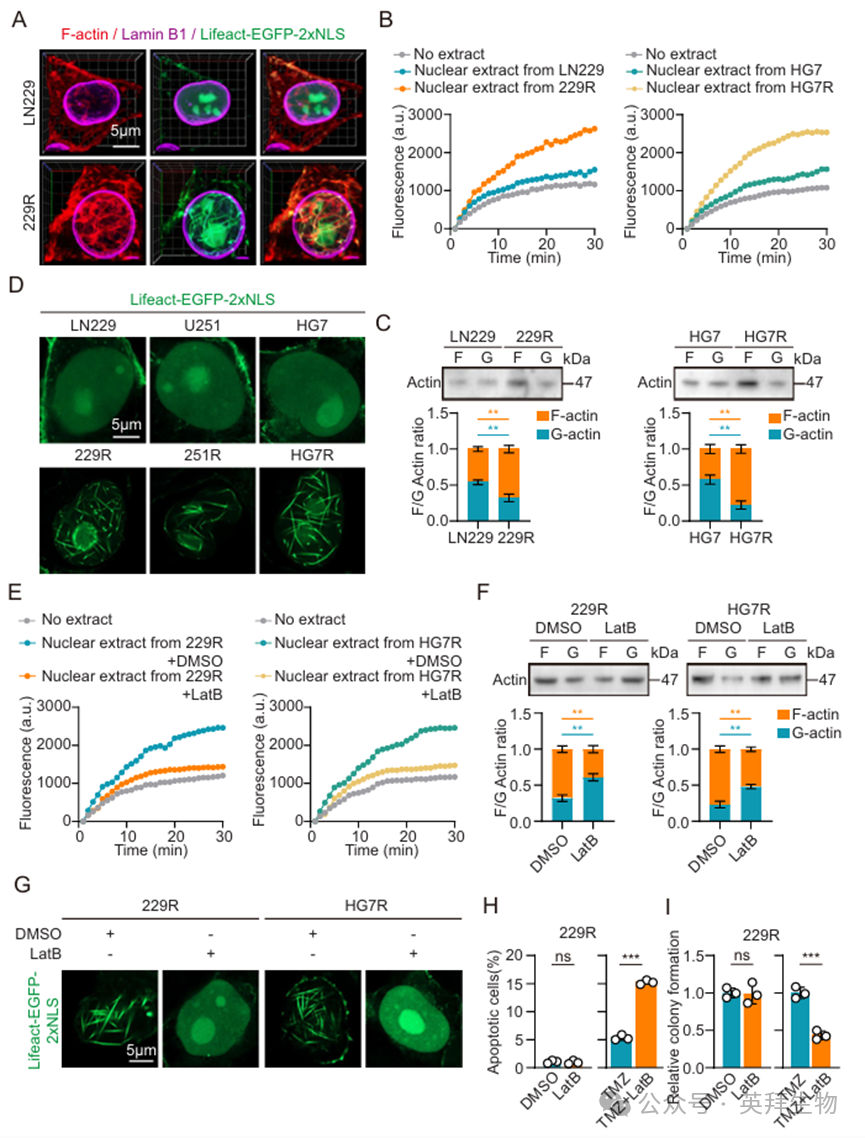
图1.核F - Actin增加促进GBM细胞对TMZ的耐药性
2.FSCN1调控核F - Actin形成促进DNA损伤修复
先前研究表明,核内F-actin与DNA双链断裂(DSBs)的修复有关。为了探究核内F-actin与GBM细胞中DNA损伤修复之间的关系,我们对TMZ耐药的GBM细胞分别进行了DMSO、LatB、TMZ以及TMZ+LatB处理。我们通过免疫荧光、彗星试验和Western blot技术评估了核内DSB损伤。结果显示,单独使用LatB处理并未在TMZ耐药性GBM细胞中诱导出显著的DNA损伤变化。然而,与仅使用TMZ处理相比,TMZ与LatB联合处理导致核内DNA损伤位点、DNA损伤尾部增加,以及DSB相关标志蛋白的积累(图2A–C;图S2A–C)。接下来,我们旨在鉴定影响TMZ耐药性GBM细胞中F-actin聚合的关键分子。在针对多种肌动蛋白调节蛋白进行初步实验后,我们聚焦于肌动蛋白束缚蛋白Fascin(FSCN1)。随后,我们通过Western blot和免疫荧光实验检测了亲代及TMZ耐药性GBM细胞核内及全细胞提取物中FSCN1的表达水平。结果显示,TMZ耐药性GBM细胞的细胞核及全蛋白水平中FSCN1表达显著升高,复发性GBM患者的组织样本中亦呈现相同趋势(图2D–F;图S2D)。此外,qRT-PCR结果显示,与亲代GBM细胞和原发性GBM患者组织相比,TMZ耐药性GBM细胞和复发性GBM患者组织中FSCN1 mRNA的表达显著增加(图S2E、F)。为了研究FSCN1表达水平与GBM细胞中核F-actin形成之间的关系,我们在TMZ耐药性GBM细胞中建立了shFSCN1细胞模型。我们通过qRT-PCR和Western blot验证了FSCN1的表达,结果显示,在shFSCN1处理的TMZ耐药性GBM细胞中,FSCN1蛋白在细胞核和总细胞提取物中的表达均降低,且FSCN1 mRNA表达水平也较低(图2G;图S2G)。核F-actin的免疫荧光染色显示,在 shFSCN1 处理的 TMZ 耐药 GBM 细胞中,核F-actin的形成受到抑制(图 2H)。为进一步探索 FSCN1 的功能作用,我们分析了 TCGA 数据集的数据,并将患者分为 FSCN1 高表达组和低表达组。基因本体论(GO)分析表明,FSCN1 的表达与 DNA 双链断裂修复和 DNA 损伤修复复合体的形成有关(图 S3A)。随后,我们通过彗星实验和Western blot评估了shFSCN1处理后的DNA损伤水平。结果显示,在TMZ治疗后,shFSCN1显著增加了TMZ耐药性GBM细胞中的DNA损伤尾迹以及DNA损伤相关标志蛋白的表达(图2I、J;图S3B、C)。
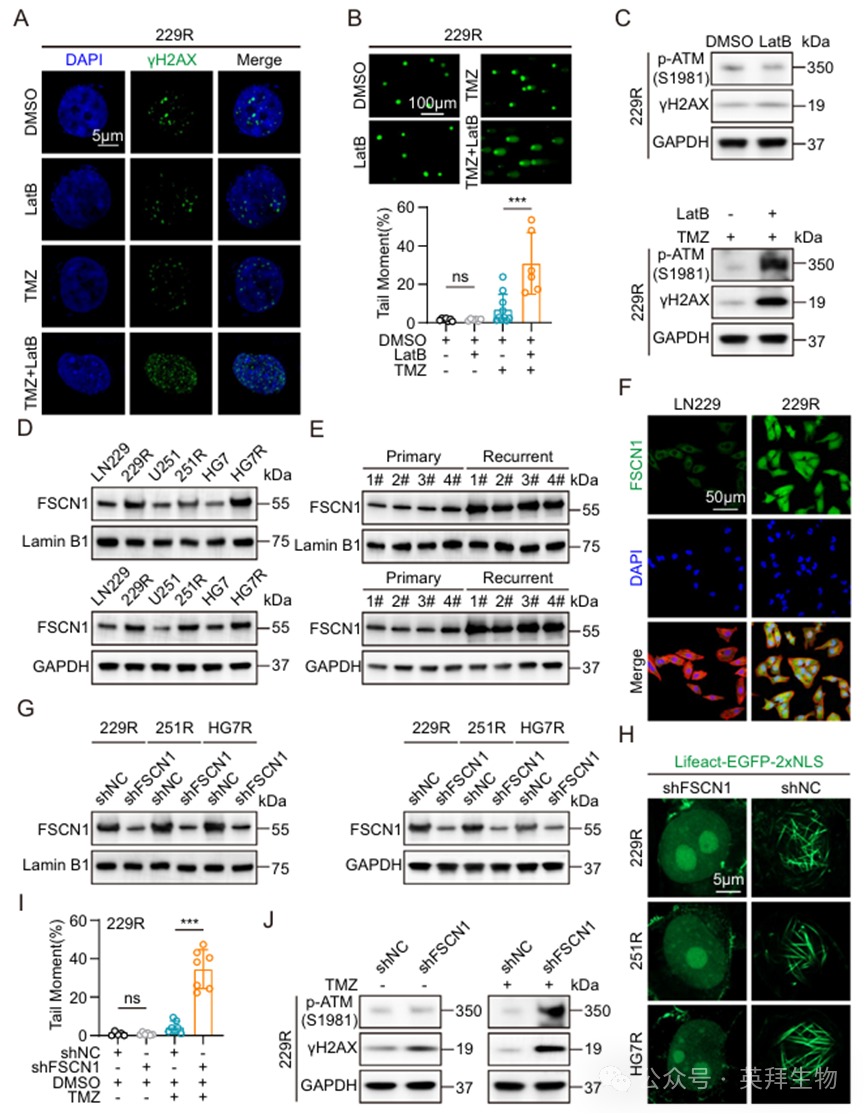
图2.FSCN1调控核F - Actin形成促进DNA损伤修复
3.YTHDC1通过m6A修饰调控FSCN1表达
基于我们之前的研究,我们利用在线工具SRAMP预测了FSCN1 mRNA上的多个高置信度和极高置信度的m6A结合位点(图3A)。为了验证这些预测,我们在GBM细胞中进行了RNA免疫沉淀(RIP)实验,使用m6A抗体和磁珠捕获与m6A结合的mRNA。纯化后,我们通过qRT-PCR检测了GBM细胞中沉淀下来的FSCN1 mRNA(图3B)。为了鉴定参与FSCN1调控的m6A甲基转移酶,我们分析了既往的测序数据集和m6A酶谱。结果显示,YTHDC1是唯一的共同因子(图S4A)。我们先前的测序数据表明,YTHDC1在TMZ耐药性GBM细胞及复发性GBM患者样本中均呈现持续下调(图S4B)。随后,我们通过RIP和沉淀实验探究了YTHDC1蛋白与FSCN1 mRNA之间的相互作用。结果表明,YTHDC1蛋白在GBM细胞中与FSCN1 mRNA发生相互作用(图3C、D;图S4C)。对公共数据库(TCGA)及临床GBM患者样本中YTHDC1与FSCN1表达的关联分析显示,两者表达呈显著负相关(图3E、F)。为进一步探讨YTHDC1蛋白对FSCN1表达的影响,我们在亲代GBM细胞(LN229、U251、HG7)中建立了s∖shYTHDC1细胞系,并在TMZ耐药GBM细胞(229R、251R、 HG7R)建立了稳定的OE-YTHDC1细胞系,并通过qRT-PCR和Western blot实验验证了YTHDC1的表达水平(图S4D、E)。随后,我们通过qRT-PCR和Western blot检测了FSCN1的表达水平,结果显示:在亲本GBM细胞中,shYTHDC1导致FSCN1表达上调;而在TMZ耐药GBM细胞中,OE-YTHDC1则抑制了FSCN1的表达(图3G;图S4F、G)。为了进一步验证 YTHDC1 蛋白与 FSCN1 mRNA 之间相互作用的特异性,我们突变了 FSCN1 mRNA 上两个高度可信的 m6A 结合位点。在 229R shFSCN1 细胞中过表达 FSCN1-wt 和 FSCN1-mut 后,我们再次过表达了 YTHDC1。Western blot 结果显示,在 229R shFSCN1 细胞中,OE-FSCN1-wt 随后 OE-YTHDC1 导致核内 FSCN1 蛋白减少,而 OE-FSCN1-mut 随后 OE-YTHDC1 则未显示核内 FSCN1 的显著变化(图 3H)。随后,我们使用放线菌素 D 抑制转录,并通过 qRT-PCR 测定了 LN229 shYTHDC1 细胞和 229R OE-YTHDC1 细胞在 0、1.5、3 和 4.5 小时时的 FSCN1 mRNA 表达水平。结果表明,随着 YTHDC1 表达的降低,FSCN1 mRNA 的衰减速率显著降低(图 3I)。
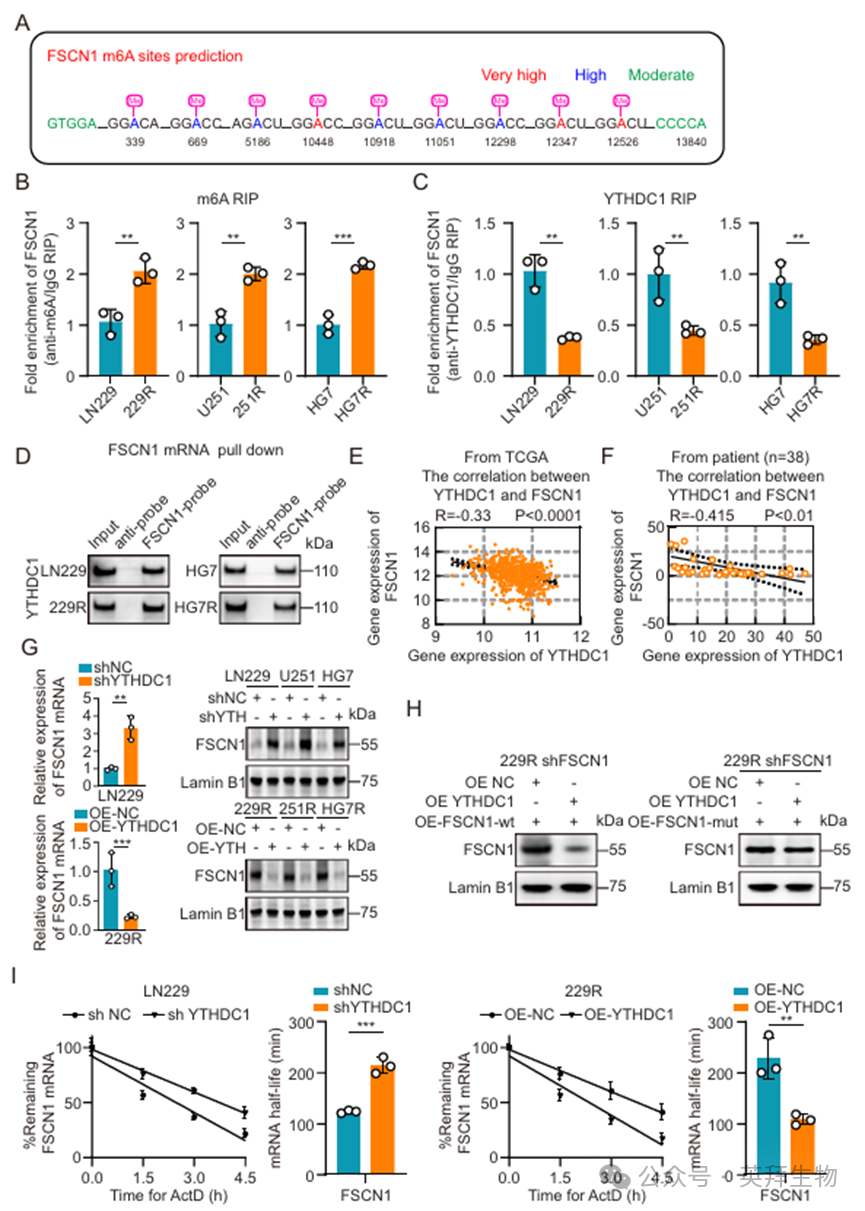
图3.YTHDC1通过m6A修饰调控FSCN1表达
4.YTHDC1在TMZ耐药的GBM细胞和复发患者中的下调与TMZ耐药相关
为进一步研究YTHDC1在GBM组织和细胞中的表达情况,我们采用qRT-PCR技术检测了27例原发性及11例复发性GBM患者肿瘤样本中YTHDC1的mRNA水平。结果显示,复发性GBM组织中YTHDC1的表达显著降低,且在4对配对样本(原发性与复发性GBM组织)中观察到了相同的结果(图4A;图S5A)。此外,qRT-PCR分析显示,与亲代细胞相比,TMZ耐药性GBM细胞中的YTHDC1表达水平显著降低(图4B;图S5B)。Western blot结果进一步证实,与原发性肿瘤和亲代细胞相比,复发性GBM组织和TMZ耐药性GBM细胞中的YTHDC1蛋白水平均显著降低(图4C、D;图S5C)。免疫组化(IHC)染色同样显示复发性GBM肿瘤中YTHDC1蛋白水平显著降低(图4E),而免疫荧光染色则显示TMZ耐药性GBM细胞中YTHDC1蛋白水平显著下降(图4F;图S5D)。为探索 YTHDC1 表达与 TMZ 耐药性之间的关系,我们分析了 TCGA 数据库中的患者数据,结果表明,较低的 YTHDC1 表达与较差的生存结果相关(图 S5E)。随后,对GBM细胞核内F-actin进行的免疫荧光染色显示:在亲代GBM细胞中,shYTHDC1促进了核内F-actin的形成,而在替莫唑胺耐药的GBM细胞中,OE-YTHDC1则抑制了核内F-actin丝的形成(图4G;图S5F)。为了确定YTHDC1在TMZ耐药性中的功能作用,我们进行了流式细胞术凋亡检测、集落形成实验和CCK8检测。结果显示,在亲本GBM细胞中,shYTHDC1在TMZ处理下可减少凋亡并增强增殖(图4H–J;图S5G–I)。接下来,我们通过在 LN229 shYTHDC1 细胞中敲低 FSCN1,以及在 229R OE-YTHDC1 细胞中过表达 FSCN1,进行了拯救实验(图 4K)。免疫荧光染色显示,shFSCN1抑制了LN229 shYTHDC1细胞中核内F-actin的形成,而OE-FSCN1则恢复了229R OE-YTHDC1细胞中核内F-actin的形成(图4L)。经 TMZ 处理后,流式细胞术凋亡检测显示,shFSCN1 恢复了 LN229 shYTHDC1 细胞对 TMZ 的敏感性,而 OE-FSCN1 则恢复了 229R OE-YTHDC1 细胞对 TMZ 的耐药性(图 4M)。
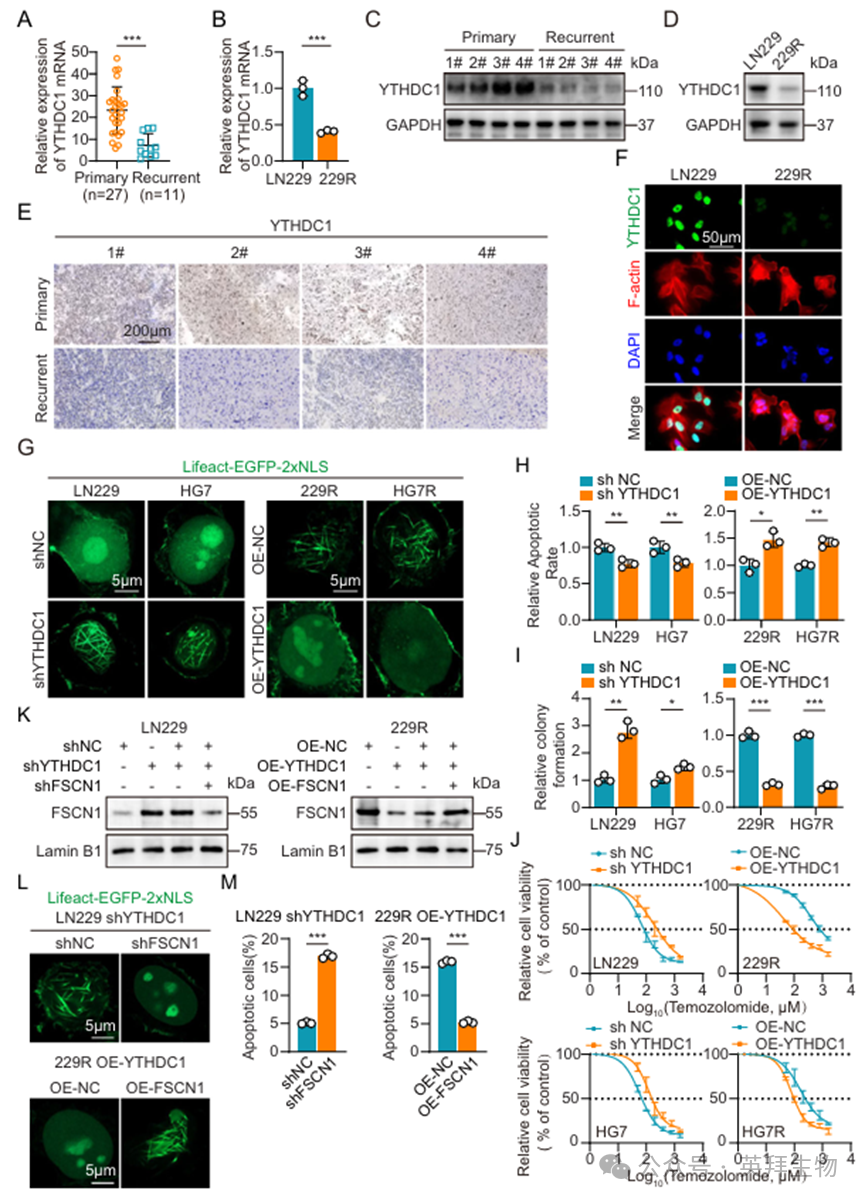
图4.YTHDC1在TMZ耐药的GBM细胞和复发患者中的下调与TMZ耐药相关
5.FSCN1 通过招募 FGD1 激活 CDC42/N-WASP/Arp2/3 轴
为探究FSCN1调节肌动蛋白丝形成和DNA DSB修复的机制,我们根据TCGA数据库中的基因集数据,将GBM患者分为FSCN1高表达组和低表达组。KEGG通路富集分析显示,“肌动蛋白细胞骨架调节”通路与我们的实验结果显著相关(图S6A)。进一步的基因集富集分析(GSEA)也证实,FSCN1的表达能够激活“肌动蛋白细胞骨架调控”通路(图S6B)。基于既往研究,我们发现FSCN1的表达可能与CDC42/N-WASP/Arp2/3信号轴的激活密切相关。接下来,我们通过 Western blot 评估了 LN229 和 229R 细胞中的 CDC42 激活水平(CDC42GTP)。结果显示,虽然 229R 细胞核提取物中的总 CDC42 水平未出现显著变化,但 CDC42GTP 水平却明显升高(图 5A)。比较 LN229 和 229R 细胞核提取物中的 N-WASP 和 Arp2/3 水平发现,它们的核蛋白水平没有显著差异。然而,通过使用N-WASP抗体进行免疫沉淀(IP)并定量分析Arp2,我们发现229R细胞核中与N-WASP结合的Arp2蛋白更多(图5B)。这些结果表明,在对TMZ耐药的GBM细胞中,CDC42/N-WASP/Arp2/3通路被激活。随后,我们对229R shFSCN1细胞的核提取物中该通路的激活情况进行了研究。Western blot和吲哚-肌动蛋白实验表明,shFSCN1显著抑制了CDC42的激活以及N-WASP和Arp2/3的结合(图5C–E)。鉴于FSCN1本身并不直接激活CDC42,我们进行了FSCN1免疫沉淀和质谱分析以鉴定其结合伙伴。筛选结果表明,GEF家族成员FGD1可与FSCN1结合并激活CDC42。Western blot分析显示,TMZ耐药性GBM细胞的细胞核中FGD1表达上调,且shFSCN1显著降低了FGD1蛋白水平(图5F、G)。免疫沉淀实验进一步证实了FSCN1与FGD1之间的相互作用(图5H)。随后,我们在LN229细胞中过表达FSCN1(OE-FSCN1),并通过Western blot和吡啶-肌动蛋白实验检测了FGD1的水平以及CDC42/N-WASP/Arp2/3通路的激活情况。结果表明,OE-FSCN1 提高了细胞核中 FGD1 的蛋白水平,并激活了 CDC42/N-WASP/Arp2/3 通路(图 5I、J)。随后,我们在LN229 OE-FSCN1细胞中敲低FGD1,进行了拯救实验。Western blot和吡啶-肌动蛋白检测显示,shFGD1抑制了LN229 OE-FSCN1细胞中核内F-actin的形成(图5K、L),这表明在缺乏FGD1的情况下,FSCN1无法独立激活CDC42/N-WASP/Arp2/3通路。为了探索 CDC42/N-WASP/Arp2/3 通路的治疗潜力,我们使用了 CDC42(ML141)和 Arp2/3(CK-666)的小分子抑制剂。免疫荧光结果显示,抑制 CDC42 或 Arp2/3 均可阻断 TMZ 耐药性 GBM 细胞中F-actin的形成(图 5M、N;图 S6C、D)。此外,我们利用遗传学方法在 229R 细胞中敲低 CDC42、N-WASP 和 Arp2,并通过 Western blot、吲哚-肌动蛋白检测和彗星实验评估了F-actin的形成和 DNA 损伤。结果表明,shCDC42、shN-WASP 和 shARP2 显著抑制了核F-actin的聚合,并增加了 TMZ 处理后的 DNA 损伤尾迹(图 S6E–G)。最后,在对TMZ耐药的GBM细胞进行shFSCN1或抑制剂ML141和CK-666处理后,免疫荧光分析显示,DNA DSB修复位点在TMZ处理后未能从异染色质区域迁移至其周边以进行同源重组(HR)修复(图5O–Q;图S6H–J)。为了探究FSCN1表达水平与细胞内HR修复事件发生频率之间的关系,我们利用DR-GFP系统对LN229 OE-NC、OE-FSCN1、229RshNC和shFSCN1细胞进行了检测。在有或无 ISceI 表达的情况下进行 TMZ 处理后,我们通过流式细胞术测定了 GFP 阳性细胞的百分比。结果表明,在 GBM 细胞中,FSCN1 表达越高,TMZ 处理下的 HR 修复频率就越高(图 5R)。综上所述,这些结果表明,FSCN1通过招募FGD1来激活CDC42/N-WASP/Arp2/3信号轴,从而促进DSB的修复,并促使DSB修复位点从异染色质区域向其周边迁移,以在GBM细胞中进行同源重组修复。
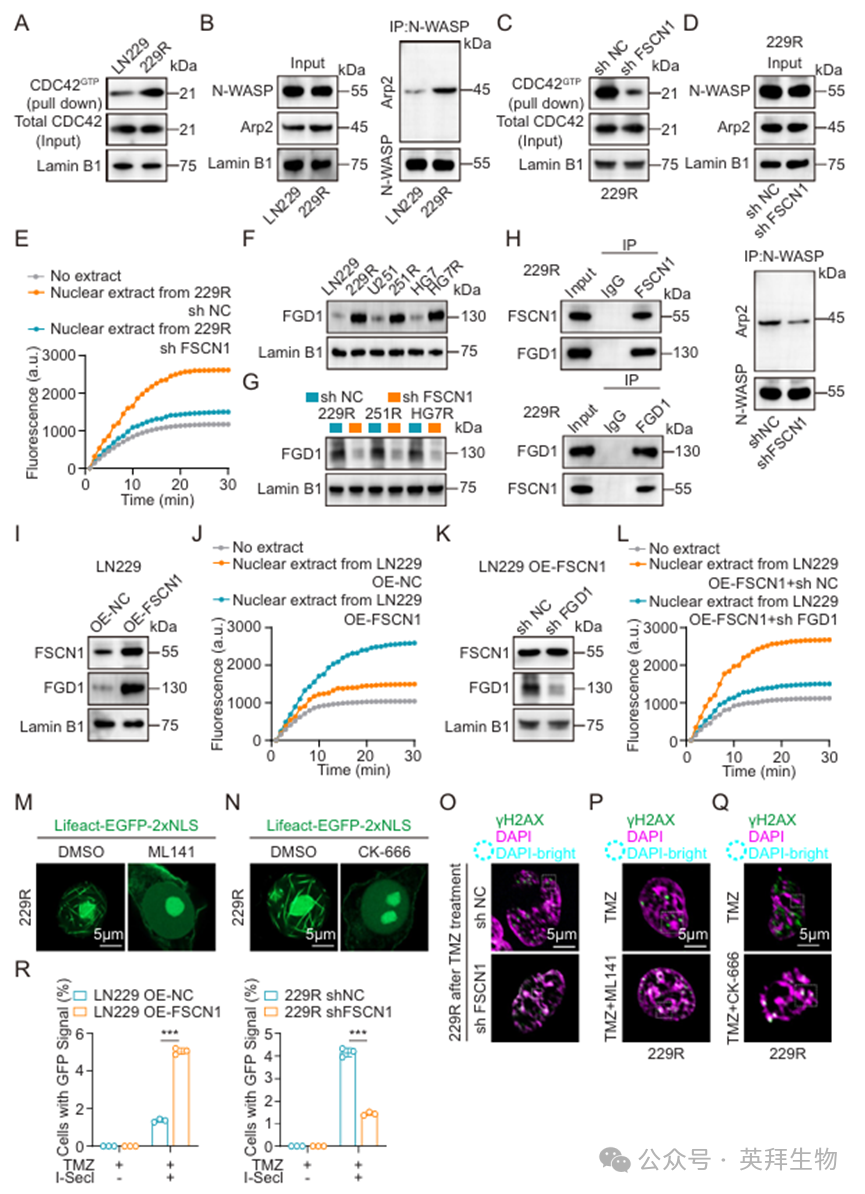
图5.FSCN1 通过招募FGD1 激活 CDC42/N-WASP/Arp2/3 轴
6.TMZ联合FSCN1抑制剂NP‐G2‐044治疗可在体内和体外逆转GBM TMZ耐药
为探索TMZ联合治疗的潜在临床应用,我们选择了目前正在进行临床试验的FSCN1抑制剂NP-G2-044,该药物主要针对其在肿瘤侵袭和转移中的作用。为验证NP-G2-044在GBM细胞中对FSCN1的特异性,我们进行了细胞热变性分析(CETSA)。Western blot结果显示,NP-G2-044显著提高了FSCN1的热稳定性(图6A)。免疫荧光染色显示,在TMZ耐药的GBM细胞中,NP-G2-044处理抑制了核内F-actin的形成(图6B)。此外,在TMZ处理条件下,NP-G2-044处理显著增加了TMZ耐药性GBM细胞中的DNA损伤位点(图6C;图S7A)。Western blot结果显示,在TMZ耐药性GBM细胞中,与单独TMZ处理相比,TMZ与NP-G2-044联合处理显著提高了DNA损伤标志蛋白的水平(图6D)。CCK8细胞存活率测定表明,TMZ与NP-G2-044的联合治疗显著抑制了TMZ耐药性GBM细胞的增殖活性(图6E;图S7B)。为评估NP-G2-044对TMZ耐药性的体内影响,我们利用TMZ耐药的229R细胞系建立了小鼠原位GBM异种移植模型。移植 7 天后,小鼠每 5 天接受 TMZ(60 毫克/千克/天)单药治疗或 TMZ 与 NP-G2-044(100 毫克/千克/天)联合治疗(图 6F)。生物发光成像显示,联合治疗有效恢复了TMZ耐药异种移植瘤对 TMZ 的敏感性(图 6G)。与对照组相比,联合治疗组的动物肿瘤体积显著缩小,生存期延长(图6H、I)。综上所述,这些结果表明,TMZ与NP-G2-044的联合应用可能是克服TMZ耐药性并提高治疗效果的一种有前景的策略。
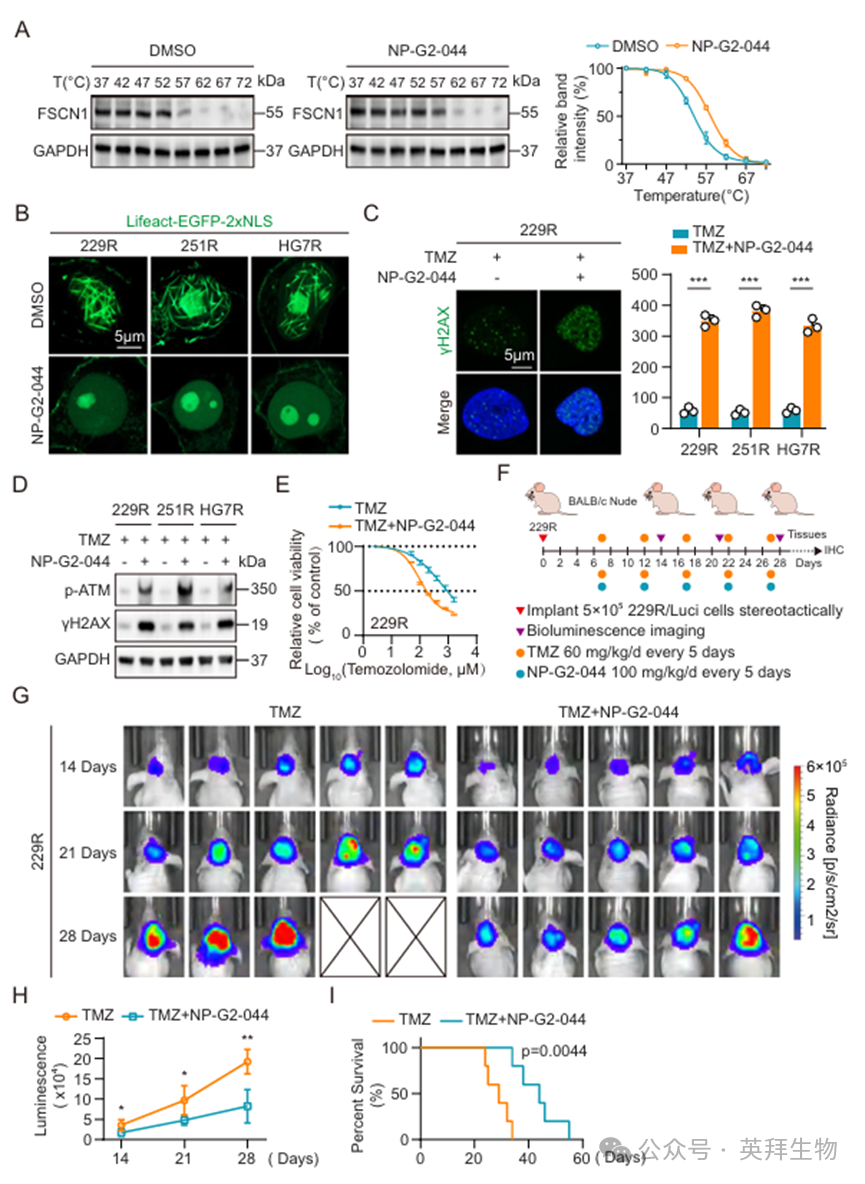
图6.TMZ联合FSCN1抑制剂NP‐G2‐044治疗可在体内和体外逆转GBM TMZ耐药
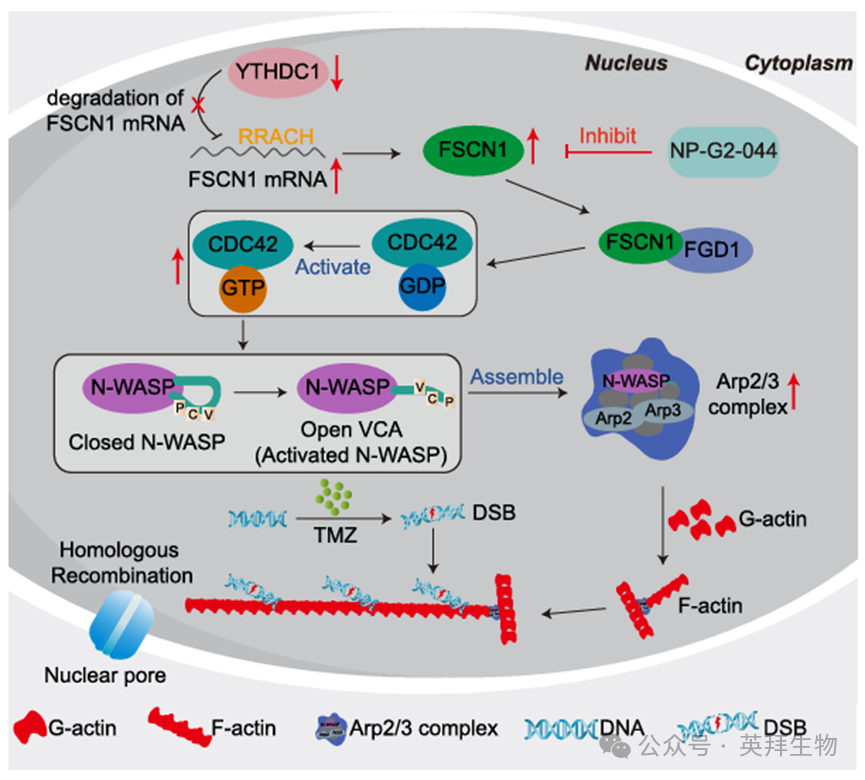
图7.在GBM中,低YTHDC1表达上调FSCN1促进核F - actin形成和促进DNA断裂修复的机制图
结论
本研究强调了核内F-actin在驱动GBM细胞对TMZ产生耐药性方面发挥的关键作用。我们证明,FSCN1通过CDC42/N-WASP/Arp2/3通路促进核内肌动蛋白的聚合,从而增强DNA损伤修复,并导致对TMZ产生耐药性。此外,YTHDC1表达的降低会进一步上调FSCN1水平,从而强化这一耐药机制。重要的是,我们的研究结果表明,将FSCN1抑制剂NP-G2-044与TMZ联合使用,为提高TMZ疗效和克服GBM耐药性提供了一种有前景的治疗策略(图7)。
参考文献
Yang M, Niu W, Wang Y, Chen P, Mu M, Zhang X, Xu B, Hu S, Niu C, Wu P. Low YTHDC1 Expression Upregulates FSCN1 to Promote Nuclear F-Actin Formation and Facilitate Double-strand DNA Breaks Repair in TMZ-Resistant Glioblastoma. Adv Sci (Weinh). 2026 Mar;13(13):e13632. doi: 10.1002/advs.202513632. Epub 2025 Dec 27. PMID: 41454694; PMCID: PMC12955858.