






IL-3 调节小胶质细胞极化并减轻创伤性脑损伤中的神经炎症
小胶质细胞在创伤性脑损伤(TBI)后的神经炎症进程中发挥着关键作用。白细胞介素-3(IL-3)作为一种重要的调节因子,已被证实参与多种疾病的发病机制,但其通过小胶质细胞对TBI后神经炎症的影响尚不明确。本研究评估了IL-3减轻TBI后小胶质细胞神经炎症的潜力。通过使用ABplex多指标流式联合分析法检测炎症因子,我们发现头痛及TBI患者的脑脊液(而非血液样本)中IL-3水平显著升高。此外,在TBI大鼠模型中,脑内给予外源性IL-3可减轻神经炎症并促进功能恢复。在机制上,我们鉴定出小胶质细胞中的过氧化还原蛋白-1(PRDX1)是IL-3的作用靶点。值得注意的是,当小胶质细胞中的PRDX1被特异性敲低后,IL-3对TBI大鼠的保护效应即被消除。总之,本实验研究表明,IL-3通过调节小胶质细胞极化来抑制神经炎症,从而发挥关键调控作用。IL-3通过IL-3R招募PRDX1,调节小胶质细胞极化,从而改善TBI大鼠的神经功能和预后。因此,IL-3可能代表一种新的TBI治疗策略。本文于2026年4月发表于《Advancedscience》, IF 14.1.
图形摘要
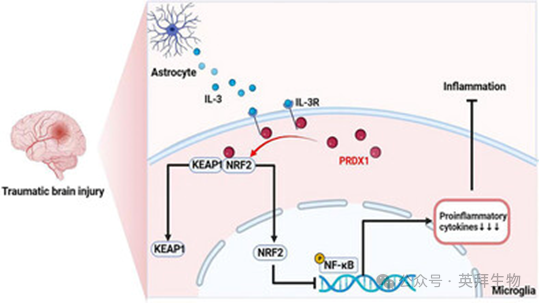
主要实验结果
1、IL-3主要由中枢神经系统分泌,并可能与TBI患者的功能恢复呈正相关
收集TBI患者(n=20)在第3、7和10天的脑脊液(CSF)和血液(BS)样本,并以头痛患者的相应样本作为阴性对照(n=20)(图1A)。ABplex多指标流式分析显示,CSF中的IL-3水平始终高于BS,并在第10天观察到显著升高(图1B)。CSF和BS中IL-1β、IL-4、IL-6和IL-8的表达水平在第3天达到峰值后下降,CSF与BS之间无显著差异。CSF中IL-10和IL-12的水平随时间保持稳定,且低于BS中的水平。TBI患者在损伤后第10天的CSF中IL-3水平显著升高。相关性分析显示,第10天CSF中IL-3水平与第90天和第180天的格拉斯哥昏迷评分(GCS)呈正相关(r = 0.7956,P = 0.0001;r = 0.8143,P = 0.0001)(图1C),而与第90天和第180天的改良Rankin量表评分(MRS)呈负相关(r = -0.8874,P = 0.0001;r = -0.7863,P = 0.0001)(图1D)。示意图展示了TBI大鼠中IL-3的来源细胞和靶细胞(图1E)。在大鼠中观察到的IL-3浓度变化趋势相似,表明IL-3主要在大脑中生成(n=11)(图1F)。免疫荧光(IF)分析显示,IL-3与星形胶质细胞标志物GFAP共定位,并且在TBI大鼠大脑晚期阶段IL-3表达显著增加(图1G)。TBI大鼠损伤后第7天脑组织切片的IF结果显示,IL-3与神经元标志物NEUN、小胶质细胞标志物IBA1或少突胶质细胞标志物MBP均无共定位。示意图展示了星形胶质细胞的诱导与收集过程。体外实验显示,在TNF-α(20ng/ml)和IL-1β(20ng/ml)刺激下,星形胶质细胞中IL-3的表达显著增加。TBI大鼠脑组织切片的IF结果显示,IL-3R与小胶质细胞标志物IBA1存在共定位,表明IL-3R主要表达于脑内小胶质细胞(图1H)。这些发现表明,IL-3由星形胶质细胞分泌并作用于脑内小胶质细胞,其高表达可能与TBI患者的功能恢复相关。
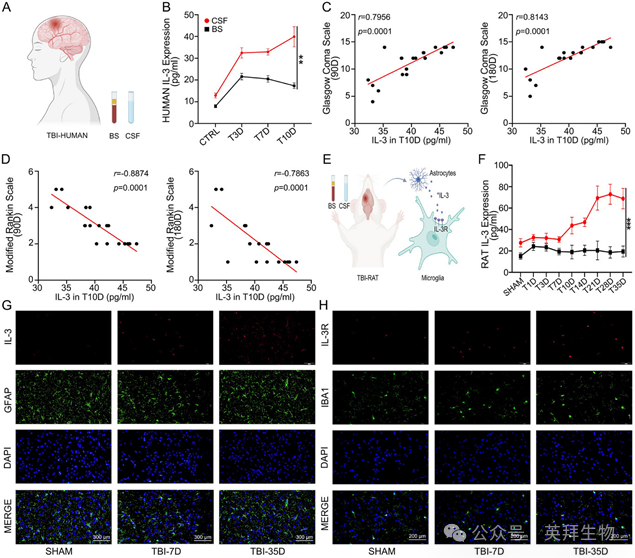
图1 TBI患者脑脊液中IL-3显著升高,并可能与功能恢复呈正相关
2、IL-3通过减轻炎症反应改善TBI大鼠的神经功能和预后
我们使用SD大鼠直接评估IL-3在TBI中的作用(n=8)。采用控制性脑皮质撞击(CCI)模型诱导TBI。在损伤后第0天和第3天,通过脑立体定位方法将重组大鼠IL-3注射至实验组大鼠左侧皮质,阴性对照组则注射生理盐水(NS)(图2A)。我们通过在健康大鼠中进行的系统剂量筛选实验(0、10、20、30µg/kg)评估了IL-3的安全性和抗炎效果。各组大鼠的存活率无显著差异(图2B)。在损伤后第35天,对不同浓度IL-3给药后的TBI大鼠心脏、肝脏、肾脏、肺和脾脏的组织损伤程度进行HE染色评估(图2C)。所有受试对象均为雄性Sprague-Dawley大鼠。各组大鼠的体重和食物消耗量保持相当。损伤后第7天对TBI大鼠进行iNOS表达的蛋白质印迹(WB)分析,结果显示20和30µg/kg剂量表现出最佳的抗炎效果(图2D、E)。对于体内实验,选择20µg/kg的IL-3浓度进行验证。在TBI前(Bf-T)以及损伤后第7、14、21、28和35天进行行为学测试,包括前肢抓力测试、倾斜平面测试、机械痛觉过敏测试、Y迷宫测试和改良神经功能缺损评分(mNSS)。结果显示,与TBI+NS组相比,接受IL-3治疗的TBI大鼠恢复速度显著更快且效果更好(图2F–K)。MRI-T2和HE染色显示,在损伤后第35天,与TBI组相比,IL-3治疗组大鼠的损伤范围显著减小(图2L–N)。尼氏染色显示,IL-3治疗促进了脑损伤区域的神经元修复(图2O)。此外,qPCR、IF和WB分析显示,IL-3治疗后iNOS显著下调(图3A–E)。同样,ELISA结果显示,IL-3治疗组大鼠脑脊液中IL-6和TNF-α等炎症因子的分泌减少(图3F、G)。在TBI后第7天,WB结果显示,IL-3治疗后p-P65显著下调,IκBα上调(图3H–J)。这些结果表明,IL-3治疗通过抑制iNOS、IL-6和TNF-α的表达来调节小胶质细胞的炎症反应,从而有效减轻炎症反应,改善TBI大鼠的神经功能和预后。
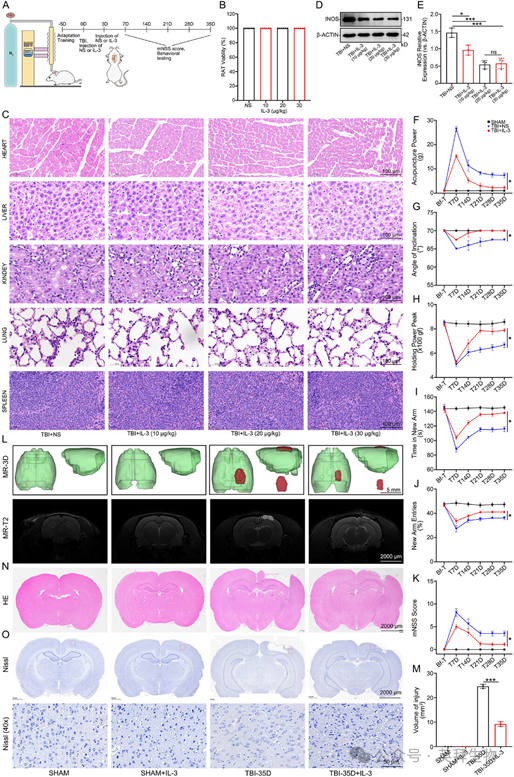
图2 IL-3改善TBI大鼠的神经功能和预后
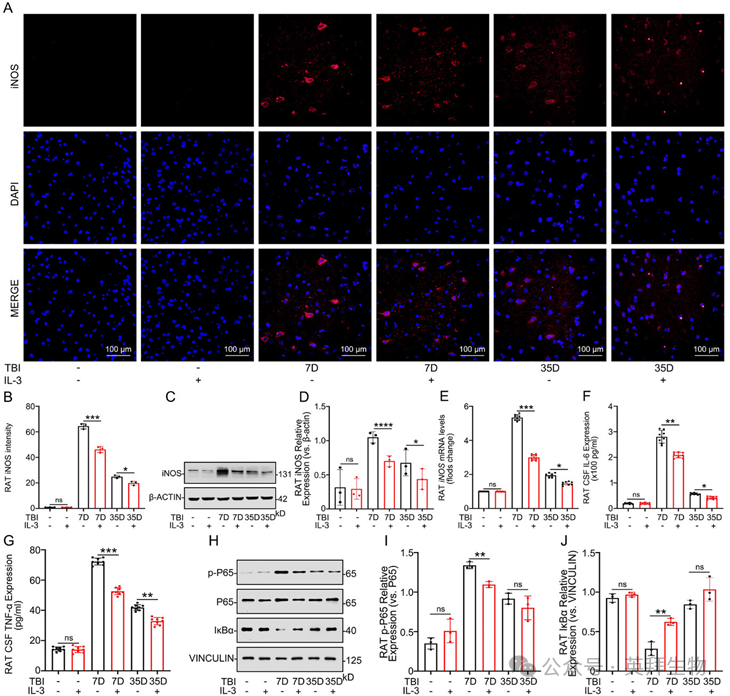
图3 IL-3调节体内炎症反应
3、IL-3调节小胶质细胞的炎症反应,并可能通过抑制体外NF-κB信号通路发挥作用
在IL-3预处理后,IF、WB和PCR分析显示原代小胶质细胞中iNOS表达下调(图4A–E)。ELISA结果显示,IL-3预处理后原代小胶质细胞中IL-6和TNF-α的分泌减少(图4F,G)。这些结果表明,IL-3在体外可抑制小胶质细胞的炎症反应。为了解IL-3影响小胶质细胞的详细机制,对经LPS+IFN-γ诱导并接受或不接受IL-3预处理的小胶质细胞进行了RNA测序(RNA-seq)(每组n = 5)。LPS+IFN-γ组与LPS+IFN-γ+IL-3组的主成分分析(PCA)显示明显的组间分离(图4H)。与LPS+IFN-γ+IL-3组相比,LPS+IFN-γ组共有2015个差异表达基因,其中934个mRNA显著上调,1081个mRNA显著下调(图4I)。KEGG富集分析揭示了多个主要通路的参与,包括NF-κB、Notch和VEGF信号通路,其中NF-κB通路显著富集(图4J)。通过IF实验确定IL-3是否能通过NF-κB信号通路作用于小胶质细胞。结果表明,LPS+IFN-γ刺激促进小胶质细胞中P65的核转位,而IL-3预处理可抑制这一过程(图4K,L)。WB结果显示,在不同时间点(0、15、30和60分钟),IL-3均可抑制P65的核转位(图4M–O)。此外,我们评估了不同时间点(0、15、30和60分钟)P65和IκBα的表达。WB结果显示,LPS+IFN-γ促进了P65的磷酸化并抑制了IκBα的表达,而IL-3预处理可干扰这些效应(图4P–R)。这些发现表明,IL-3可能通过NF-κB通路抑制iNOS、IL-6和TNF-α的表达,从而抑制小胶质细胞的炎症反应。
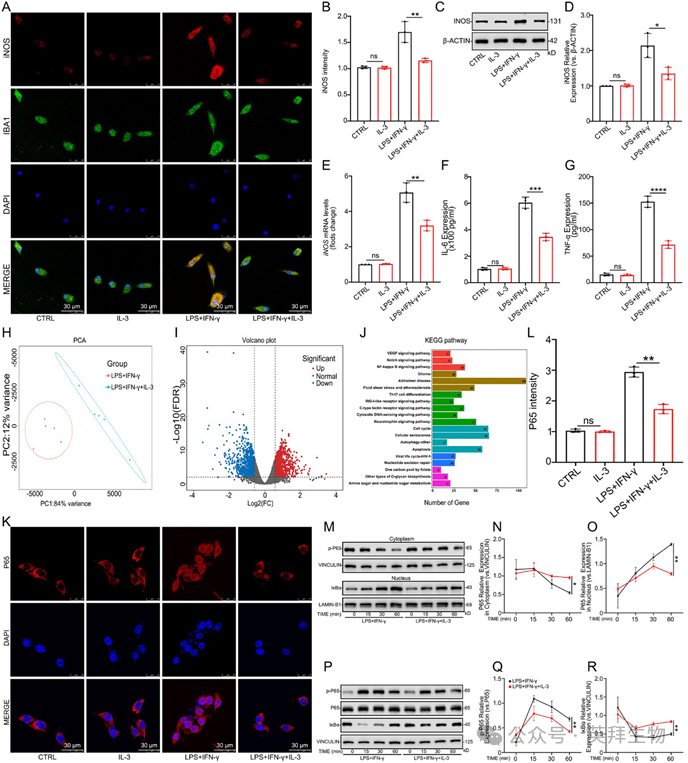
图4 IL-3通过抑制体外NF-κB信号通路抑制小胶质细胞的炎症反应
4、PRDX1作为小胶质细胞中IL-3的下游靶点
为了鉴定小胶质细胞中IL-3的下游靶点,我们使用GST下拉实验(GST-pulldown)分离了经IL-3处理后与IL-3R结合的蛋白质(图5A)。通过液相色谱-质谱联用(LC-MS)对分离出的蛋白质进行分析,结果显示PRDX1与神经系统炎症密切相关(图5B,C)。免疫共沉淀(Co-IP)实验证实,IL-3促进了IL-3R与PRDX1的结合(图5D–F)。IF结果进一步显示,IL-3促进了PRDX1向细胞膜的转位,从而增强其与IL-3R的结合(图5G)。这些结果表明,PRDX1可能是小胶质细胞中IL-3的关键靶点。
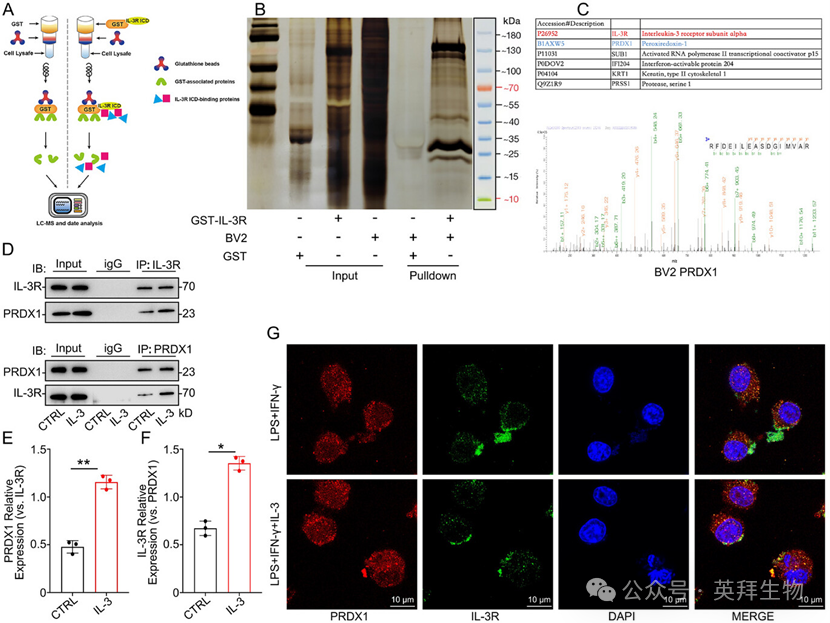
图5 PRDX1作为小胶质细胞中IL-3的下游靶点
5、IL-3通过PRDX1减轻TBI大鼠的神经炎症
为了研究PRDX1在IL-3效应中的作用,通过脑立体定位技术将腺相关病毒AAV9-IBA1-PRDX1或对照AAV载体靶向注射至大鼠左侧皮质,以敲低PRDX1(图6A)。21天后,通过AAV携带的GFP表达确认转染效率。脑组织的WB分析证实了PRDX1的有效敲低。在PRDX1敲低后,通过立体定位注射将重组大鼠IL-3给予TBI大鼠的左侧皮质。在AAV-NC组中,IL-3治疗显著改善了前肢抓力测试、倾斜平面测试、机械痛觉过敏测试、Y迷宫测试和mNSS的表现;而在AAV-PRDX1组大鼠中,这种改善不显著(图6B–G)。TBI后大鼠的MRI-T2成像和HE染色结果显示,IL-3治疗减少了注射AAV-NC大鼠的损伤面积,但在AAV-PRDX1大鼠中未见显著减少(图6H–J)。脑组织的尼氏染色结果也支持这些发现(图6K)。对损伤部位的qPCR、WB和IF分析表明,AAV-PRDX1降低了IL-3抑制iNOS表达的能力(图6L–P)。同样,在存在AAV-PRDX1的情况下,IL-3对炎症因子IL-6和TNF-α的抑制作用也受损(图6Q,R)。这些结果表明,IL-3可能通过PRDX1介导的iNOS、IL-6和TNF-α表达下调,从而减轻TBI大鼠的神经炎症并可能促进功能恢复。
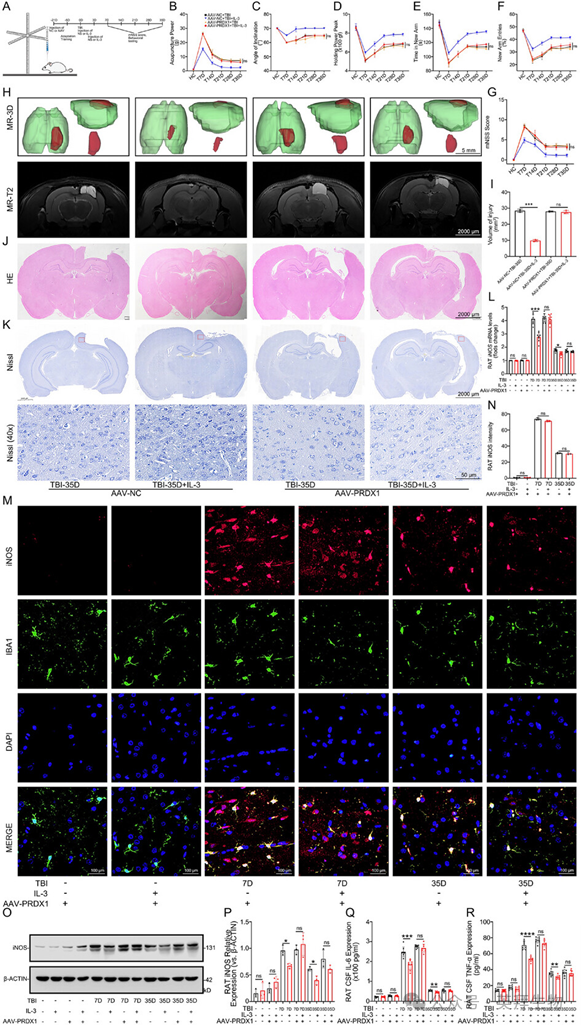
图6 IL-3通过PRDX1减轻TBI后的神经炎症
6、IL-3通过PRDX1调节NF-κB通路从而调控小胶质细胞炎症反应
为了进一步研究PRDX1在IL-3介导的小胶质细胞效应中的作用,使用sg-PRDX1(sgRNA)转染小胶质细胞以敲除PRDX1。IF和WB分析显示,sgRNA转染后PRDX1表达显著降低(图7A–C)。后续评估显示,在PRDX1敲除条件下,经IL-3处理与未处理的小胶质细胞之间iNOS表达无显著差异(图7D–G)。ELISA结果显示,在PRDX1敲除条件下,经LPS+IFN-γ刺激时,IL-3预处理组与未预处理组的小胶质细胞之间IL-6和TNF-α水平无显著差异(图7H,I)。此外,在P65磷酸化、IκBα表达或P65核转位方面也未观察到显著差异(图7J–M)。这些结果表明,IL-3通过PRDX1介导的NF-κB通路调控,抑制iNOS、IL-6和TNF-α的表达,从而调节小胶质细胞炎症反应。
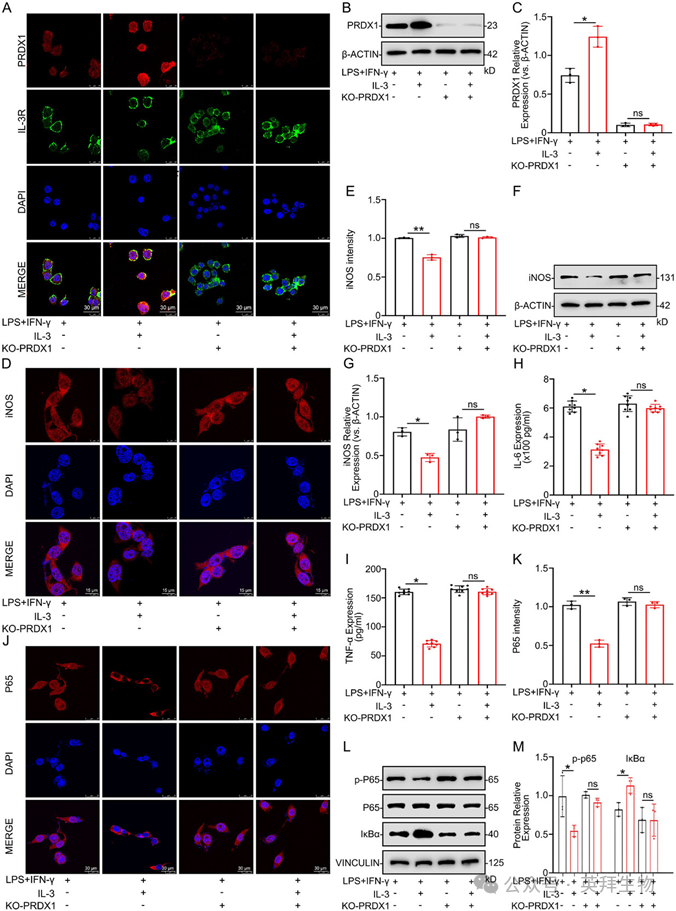
图7 IL-3作用于PRDX1以调节NF-κB通路并抑制小胶质细胞的炎症反应
7、KEAP1-NRF2-HO-1信号通路作为IL-3通过PRDX1调节神经炎症的关键机制
示意图展示了IL-3通过PRDX1调节KEAP1-NRF2-HO-1信号通路来调控神经炎症(图8A)。为了阐明KEAP1-NRF2-HO-1信号通路在IL-3作用于小胶质细胞过程中的参与情况,进行了Co-IP实验。结果显示,IL-3促进了小胶质细胞中KEAP1与NRF2的解离(图8B–D)。IF和WB分析显示,IL-3诱导了NRF2,并促进其转位进入细胞核(图8E–I)。WB结果显示,IL-3处理未改变KEAP1的表达,但显著激活了NRF2和HO-1的表达(图8J–M)。我们进一步表征了TBI后大鼠脑组织中NRF2的时间表达谱。WB结果显示,IL-3处理显著上调了NRF2的表达。在PRDX1敲除的小胶质细胞中,Co-IP分析显示IL-3无法促进KEAP1-NRF2的解离,表明PRDX1对此相互作用至关重要(图8N–P)。WB结果显示,在PRDX1敲除的小胶质细胞中,无论是否进行IL-3预处理,在LPS+IFN-γ刺激下NRF2和HO-1水平均未见显著升高(图8Q–S)。这些结果证实,IL-3通过PRDX1抑制iNOS、IL-6和TNF-α以调节小胶质细胞炎症反应,这一过程可能涉及KEAP1-NRF2-HO-1信号通路。
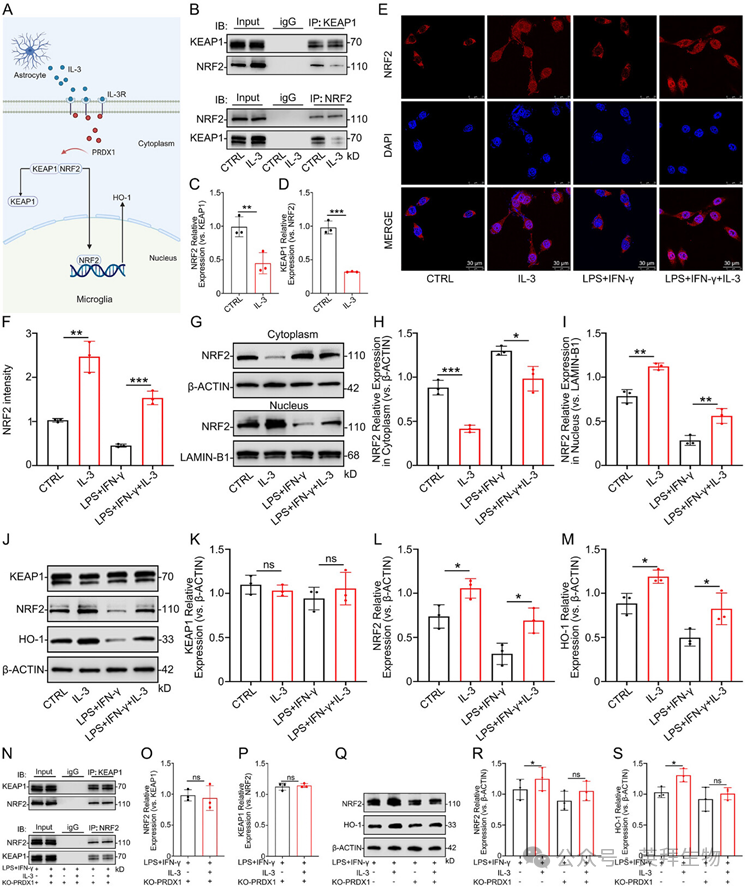
图8 KEAP1-NRF2-HO-1信号通路介导IL-3通过PRDX1调节神经炎症的作用
8、IL-3通过NRF2调节NF-κB通路以抑制小胶质细胞的炎症反应
示意图展示了IL-3通过NRF2调节NF-κB通路从而抑制小胶质细胞炎症反应(图9A)。为了进一步验证KEAP1-NRF2-HO-1通路参与IL-3调节小胶质细胞炎症的假说,使用特异性NRF2抑制剂ML385抑制NRF2。WB结果证实了NRF2的有效抑制(图9B,C)。IF和WB分析显示,抑制NRF2后,IL-3无法抑制iNOS的表达(图9D–G)。分子水平验证证实,NRF2是IL-3调节NF-κB通路中的关键转录因子,影响炎症因子IL-6和TNF-α的释放(图9H,I)。ML385抵消了IL-3对P65核转位的作用(图9J–N)。此外,WB结果显示,IL-3抑制P65磷酸化并促进IκBα表达的能力因NRF2抑制而被破坏(图9O–Q)。这些结果表明,IL-3通过PRDX1抑制小胶质细胞的炎症反应,这一过程可能涉及KEAP1-NRF2-HO-1信号通路。
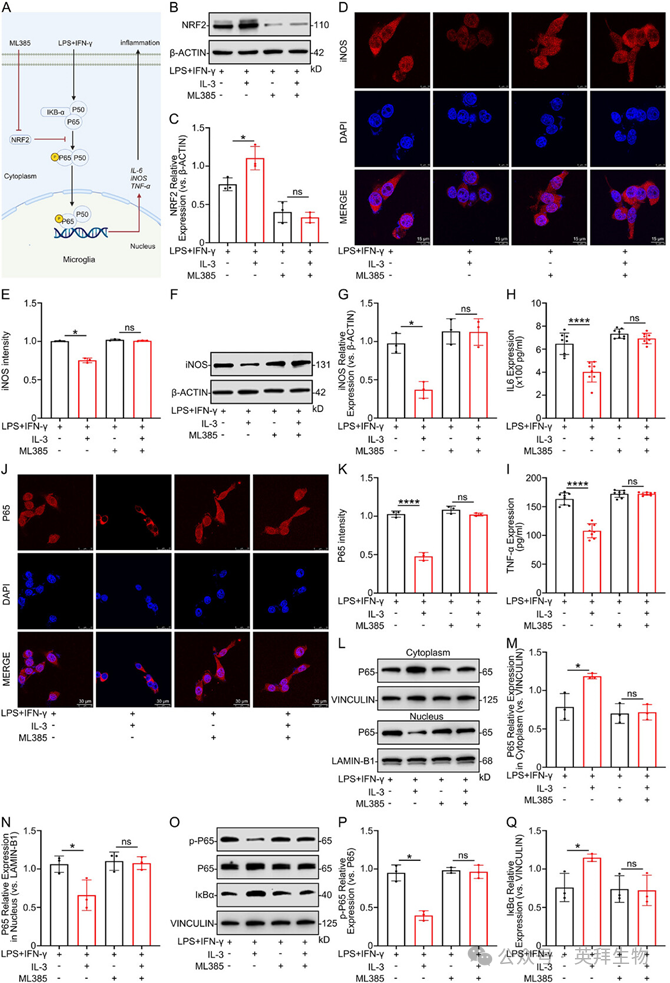
图9 IL-3通过NRF2调节NF-κB通路以抑制小胶质细胞的炎症反应
总结
总之,本研究揭示了IL-3在调节小胶质细胞极化中的关键作用。我们发现,IL-3通过IL-3R招募PRDX1,调节小胶质细胞极化,从而改善TBI大鼠的神经功能和预后。我们的研究结果证实了PRDX1在IL-3介导的KEAP1-NRF2-HO-1/NF-κB通路调节中的关键作用。本研究为TBI治疗提供了新的见解,加深了对TBI机制的理解,并为创伤性脑损伤的临床管理提供了新视角。这些发现可能为开发TBI后神经功能修复与再生的新治疗策略提供依据。
参考文献:
Huang N, Zhang Q, Chen Y, Yu D, Liu R, Kang J, Fang X, Zhang Y, Bian H, Zhao Y, Yin Y, Zhang C, Jia Y, Chen Q, Fang Y, Li S, Li F, Jin Z, Ning B. IL-3 Modulates Microglia Polarization and Attenuates Neuroinflammation in Traumatic Brain Injury. Adv Sci (Weinh). 2026 Mar 31:e04511. doi: 10.1002/advs.202504511. Epub ahead of print. PMID: 41915872.