






从代谢重编程到铁死亡:PDK4驱动乳酸累积及LPCAT2乳酸化,经STAT1/SLC7A11轴加剧脓毒症肺损伤
肺组织中糖酵解活性的升高是脓毒症诱导的急性肺损伤(SI-ALI)的标志性特征,但糖酵解重编程及乳酸衍生的蛋白质修饰在损伤上皮细胞中的作用尚不明确。在本研究中,我们发现PDK4驱动的糖酵解重编程会促进SI-ALI期间肺组织中乳酸的过量产生。从机制上讲,上皮细胞中的AARS1会选择性地增强LPCAT2蛋白K375位点的乳酸化修饰,从而抑制STAT1的乙酰化,并促进STAT1的磷酸化、核转位以及对SLC7A11的转录抑制。这一级联反应最终诱导上皮细胞发生铁死亡。药理学抑制PDK4可减轻乳酸积聚和LPCAT2的乳酸化,从而恢复STAT1的乙酰化及SLC7A11的表达。此外,敲低AARS1或突变LPCAT2-K375乳酸化位点,可在体外及败血症小鼠模型中逆转STAT1介导的SLC7A11抑制作用,并减轻铁死亡。我们的研究结果表明,PDK4表达升高是败血症期间肺组织乳酸生成增加的关键因素,并确立了驱动SI-ALI中上皮细胞铁死亡的新型LPCAT2-K375/STAT1/SLC7A11轴,突显了代谢重编程、翻译后修饰(PTM)与铁死亡之间的相互作用。针对PDK4或LPCAT2的乳酸化进行干预,可能为SI-ALI提供治疗潜力。在SI-ALI中,PDK4过度活化会导致上皮细胞产生过量乳酸,从而触发AARS1/HDAC9介导的LPCAT2乳酸化修饰。这种修饰既抑制STAT1的乙酰化,又增强其磷酸化,进而驱动STAT1向细胞核转位,并导致SLC7A11转录下调。由此导致的谷胱甘肽合成缺陷会促进铁死亡,从而加剧SI-ALI的进展。该研究于2026年3月发表在《Cell Death and Differentiation》,IF:15.4。
技术路线

主要研究结果:
1.乳酸会加重SI-ALI
血浆乳酸水平升高在临床上与脓毒症患者的病情恶化和多器官功能障碍密切相关。我们的研究发现,SI-ALI患者体内的乳酸水平显著高于健康对照组(图 1A)。我们测量了炎症因子(IL-1β、IL-6 和 TNF-α)的血浆水平,并使用动脉血氧分压(PaO2)/吸入氧分数(FiO2)比值来评估肺损伤的严重程度。PaO2/FiO2 比值是根据入院 24 小时内符合脓毒症 3 期标准的脓毒症患者的初始动脉血气分析结果计算得出的,使用的是 PaO2 和 FiO2 的值。该比值是评估 SI-ALI 的关键基准。我们的分析显示,血浆乳酸水平与炎症因子浓度之间存在正相关关系(图 1B - D),这表明乳酸水平升高可能加剧全身炎症。此外,乳酸水平升高的 SI-ALI 患者肺功能明显更差,表现为更低的 PaO2/FiO2 比值(图 1E)。
在 CLP 小鼠中,乳酸水平显著高于假手术组(图 1F、G),并且肺部损伤也更为严重。此外,在这些小鼠中,血浆乳酸浓度与炎症因子浓度之间存在正相关关系(图 1H–J)。为进一步探究乳酸与 SI-ALI 之间的关系,使用了2-DG(一种能降低乳酸生成的泛糖酵解抑制剂)对 CLP 组和假手术组的小鼠进行治疗。相关绘图材料由 BioGDP.com 提供(图 1K)。这种治疗显著降低了炎症因子水平(图 1L–N),降低了肺湿/干重比,并减轻了急性肺损伤评分(图 1O–Q)。这些结果表明,抑制乳酸生成能够有效减轻全身炎症并缓解 SI-ALI。
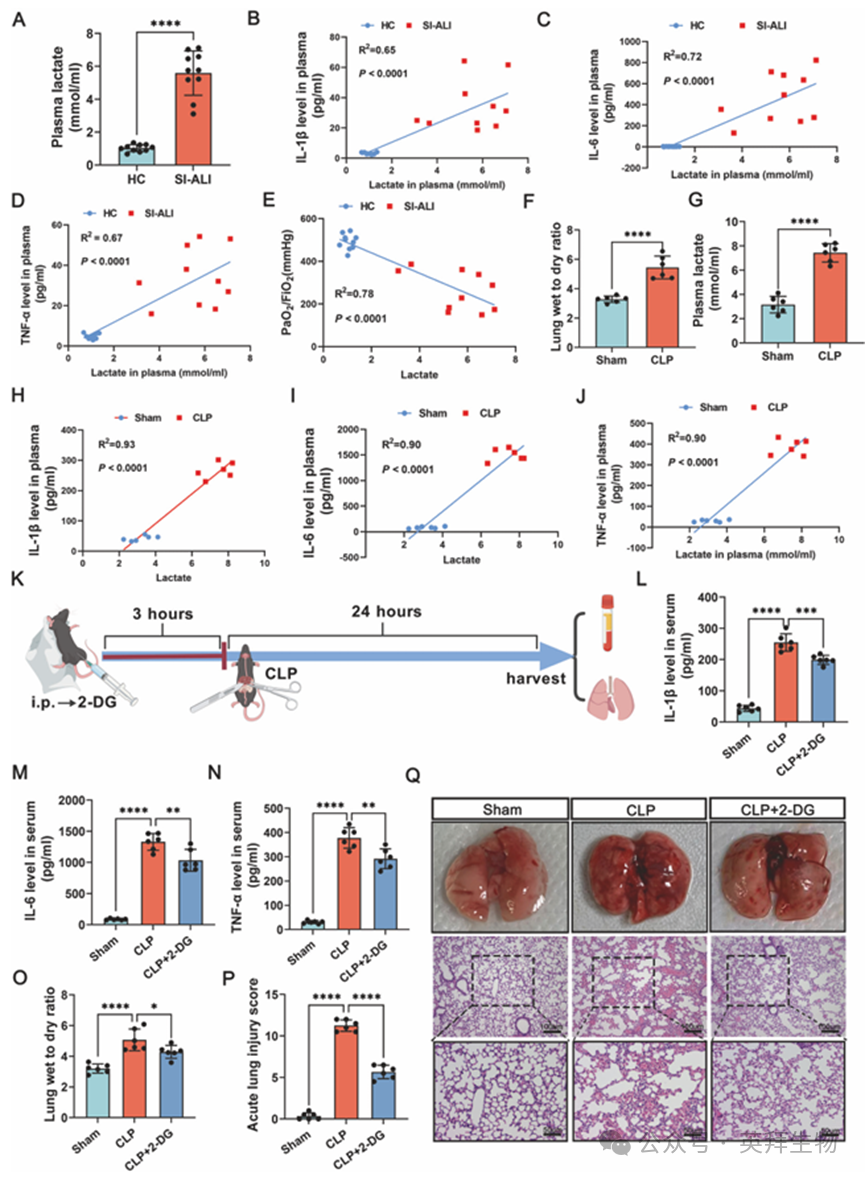
图1.抑制乳酸生成可缓解SI-ALI
2.PDK4依赖性糖酵解重编程促进SI-ALI中的乳酸积聚
乳酸生成的主要途径包括糖酵解和丙酮酸代谢,其中关键酶包括乳酸脱氢酶(LDHA/LDHB)、丙酮酸脱氢酶激酶(PDK1-4)[37]以及单羧酸转运蛋白(MCT1-4)等。在蛋白质组学分析中,热图显示 PDK4 是唯一在与乳酸相关的表达中具有统计学显著上调作用的蛋白质(图 2A、B)。PDK4-in 可阻断 PDK4 使其重新激活丙酮酸脱氢酶(PDH),将丙酮酸转化为乙酰辅酶 A,增强三羧酸循环和 ATP 生成,同时减少乳酸生成(图 2C)。使用 PDK4-in 处理显著提高了 CLP 小鼠的存活率(图 2D、E),同时减少了脓毒症肺组织中的乳酸积累和 PDK4 表达(图 2F-11)。此外,PDK4-in 治疗显著降低了 CLP 小鼠的肺湿/干重比,并减轻了急性肺损伤评分(图 2J-11)。
在体外研究中,我们发现脂多糖(LPS)能够显著增加乳酸生成量,而 PDK4 抑制剂则能显著减轻乳酸积累,并缓解乳酸引起的细胞损伤和死亡(图 2M - O)。此外,PDK4 抑制剂抑制了糖酵解代谢的重新编程,这从恢复的 ATP 水平中得以体现(图 2P)。还通过 ECAR 和 OCR 来分别量化糖酵解和线粒体呼吸作用。LPS 促进的糖酵解和抑制的线粒体呼吸作用部分被 PDK4 抑制所逆转(图 S1A - D),而 PDK4 抑制剂处理显著降低了 LPS 上调的 PDK4 表达(图 2Q,R)。综上所述,这些发现表明 PDK4 可能在 SI-ALI 中的乳酸生成过程中起关键作用,驱动代谢重编程,并促进疾病进展。
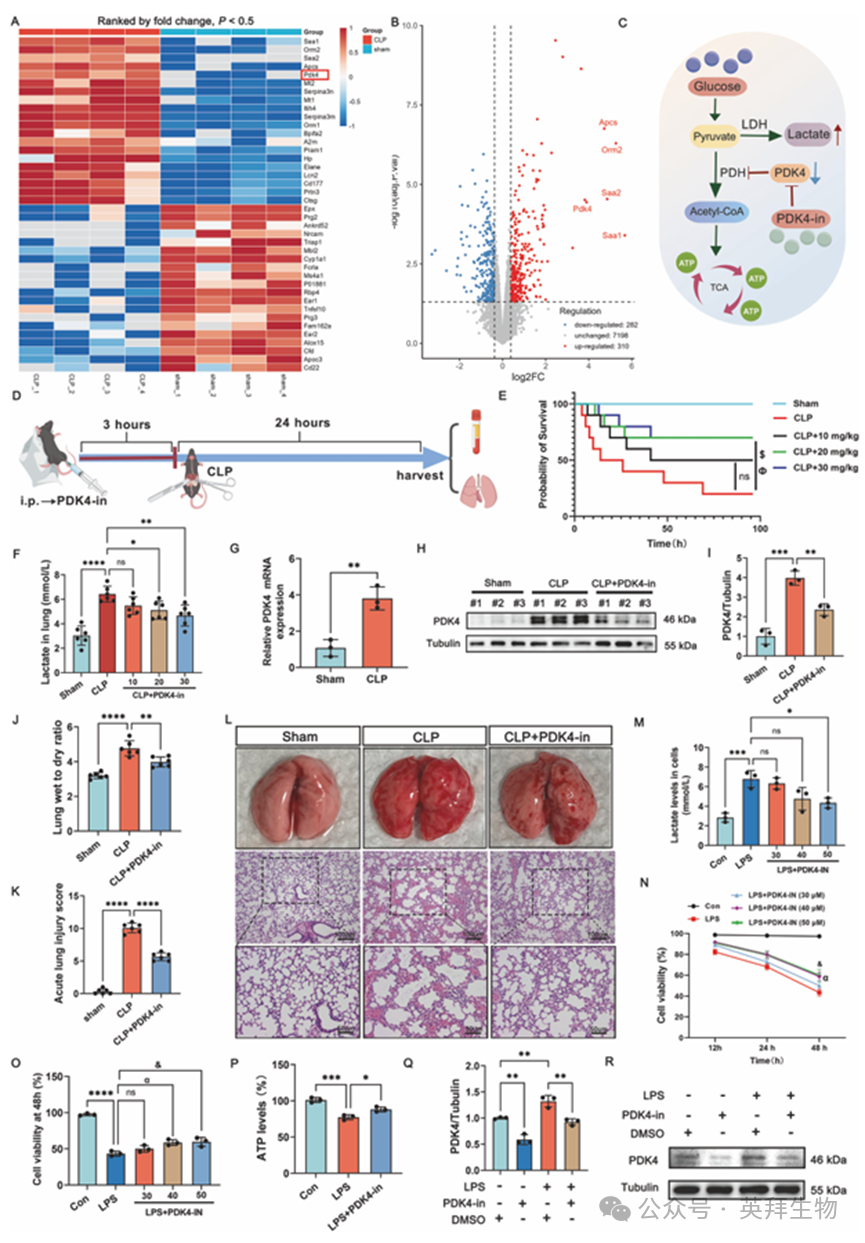
图2.PDK4表达水平升高在败血症期间肺组织乳酸过度积聚的过程中起着关键作用
3.LPCAT2-K375的乳酸化可能在SI-ALI的发病机制中起重要作用
为在 SI-ALI 组织中鉴定非组蛋白乳酸化蛋白,我们进行了乳酸化蛋白质组测序,发现了 911 种乳酸化蛋白(其中 283 个处于上调状态,807 个处于下调状态)(图 3A - E)。这种广泛的分析突显了乳酸化现象在 SI-ALI 情境中的普遍性。
在这些蛋白质中,热图分析显示 LPCAT2-K375 在 SI-ALI 中显著上调(图 3F)。此前的研究表明,LPCAT2 主要参与铁死亡进程[39, 40]。GO 和 KEGG 分析显示,在 SI-ALI 中的非组蛋白乳酸化修饰显著影响与铁死亡相关的通路(图 3G、H,补充表 3)。测序在 LPCAT2 中鉴定出两个潜在的乳化位点,但只有 K375 显示出显著的乳酸化现象,并且在各种物种中都保持一致(图 3I - K)。鉴于乳酸化分析显示在 SI-ALI 组织中 LPCAT2-K375 的乳化上调显著,这表明它是加剧肺损伤的关键作用之一。
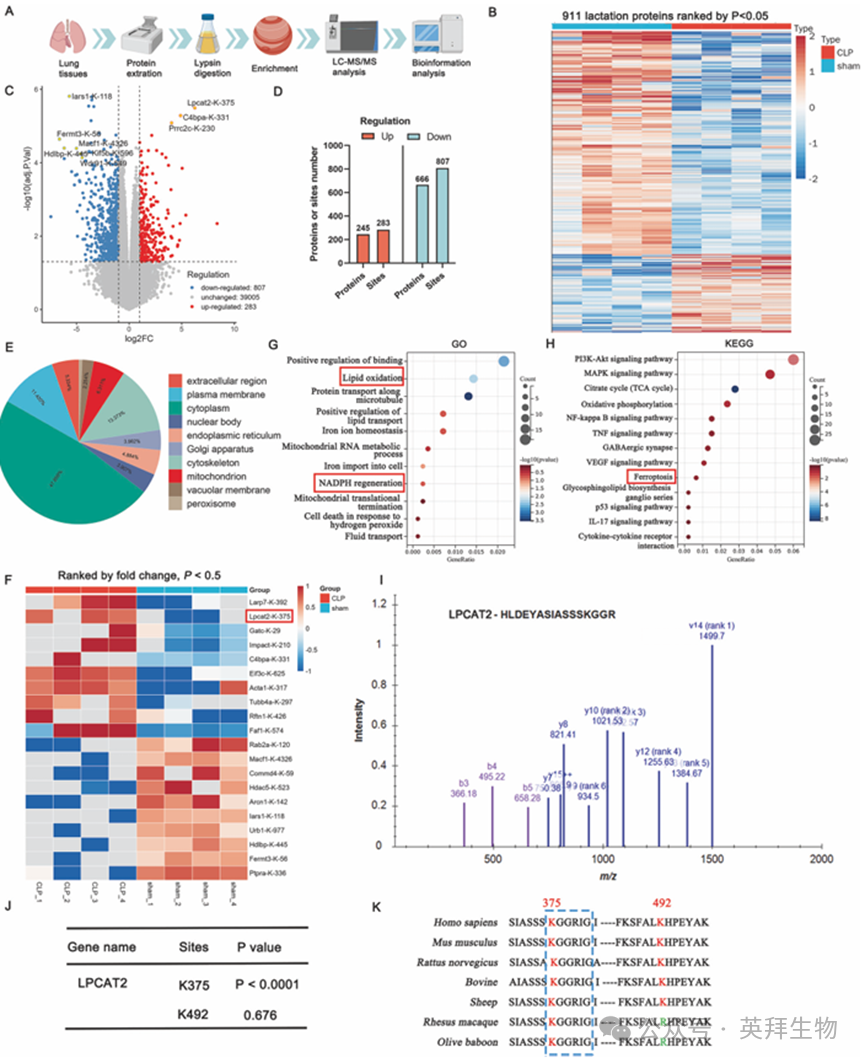
图3.LPCAT2乳酸化水平升高是SI-ALI的关键因素之一
4.SI-ALI患者LPCAT2-K375乳酸化水平显著升高
体内实验中,WB结果显示,SI-ALI 组组织中的乳酸化水平显著高于假手术组(图 4A、B)。共免疫沉淀(Co-IP)和 WB 结果显示,CLP 组中乳酸化的 LPCAT2-K375 水平上调(图 4C、D)。免疫荧光染色结果也表明,SI-ALI 组组织中 LPCAT2-K375 的表达显著增加,进一步支持其在 SI-ALI 发病机制中的潜在作用(图 4E、F)。
体外研究显示,用乳酸处理后,BEAS-2B 细胞中的乳酸化水平显著升高(图 4G、H)。共免疫沉淀实验证实了 BEAS-2B 细胞中存在乳酸化现象(图 4I、图 S4A)。此外,乳酸处理上调了 LPCAT2-K375 的表达(图 4J、K),这一结果也通过免疫荧光染色得到了验证(图 4L、M)。这些发现共同表明,乳酸诱导的 LPCAT2-K375 乳酸化促进了 SI-ALI 的发病机制,突显了其作为治疗靶点的潜力。
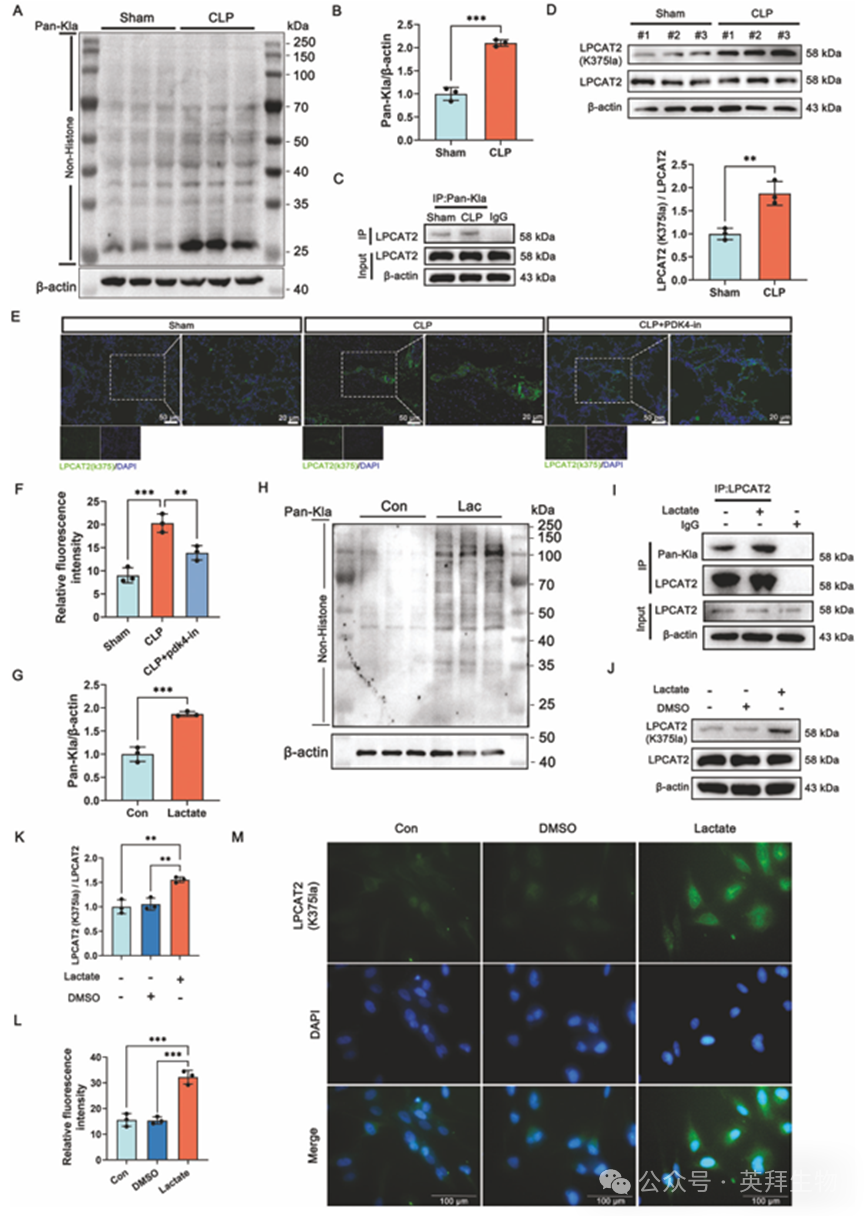
图4.乳酸可上调LPCAT2-K375的乳酸化
5.乳酸通过抑制 SLC7A11 加剧上皮细胞铁死亡
为验证乳酸和铁死亡会加重 SI-ALI 病症,我们用乳酸以及新型铁死亡抑制剂 UAMC-3203 对 CLP 小鼠进行治疗。结果表明,乳酸加剧了 SI-ALI 中的铁死亡,而 UAMC-3203 则减轻了 SI-ALI 症状(图 S2A–F)。在高乳酸条件下,SLC7A11 的水平降低,但在 PDK4-in 处理后得以恢复(图 5A–C)。CLP 组织中的线粒体功能障碍在 PDK4-in 处理后也得到了改善(图 5D),这还显著减少了脂质过氧化物的积累(图 5D–F)。这些发现表明,抑制 PDK4 可以减轻铁死亡。
体外实验表明,乳酸显著抑制了 SLC7A11 的表达(图 5G、H),而 PDK4-抑制剂的处理则逆转了这一效应(图 5I、J)。铁死亡抑制剂还增强了在高乳酸条件下 BEAS-2B 细胞的存活率(图 5K),这表明铁死亡在高乳酸环境中发生。这一现象在 MLE12 细胞中也有体现:我们发现乳酸能够上调 MLE12 细胞中脂质过氧化的水平,而铁死亡抑制剂 fer-1 则能够下调其水平(图 S3A–D)。此外,PDK4-抑制剂处理在 LPS 组中提高了 GSH 水平,并降低了 MDA/ROS 水平(图 5L–N)。综上所述,这些结果表明 LPCAT2 通过调节 SLC7A11 的表达来介导铁死亡,并且 PDK4-抑制剂能够通过恢复 SLC7A11 的表达有效地减轻铁死亡。这些发现突显了针对 PDK4 在重症急性肺损伤中的治疗潜力。
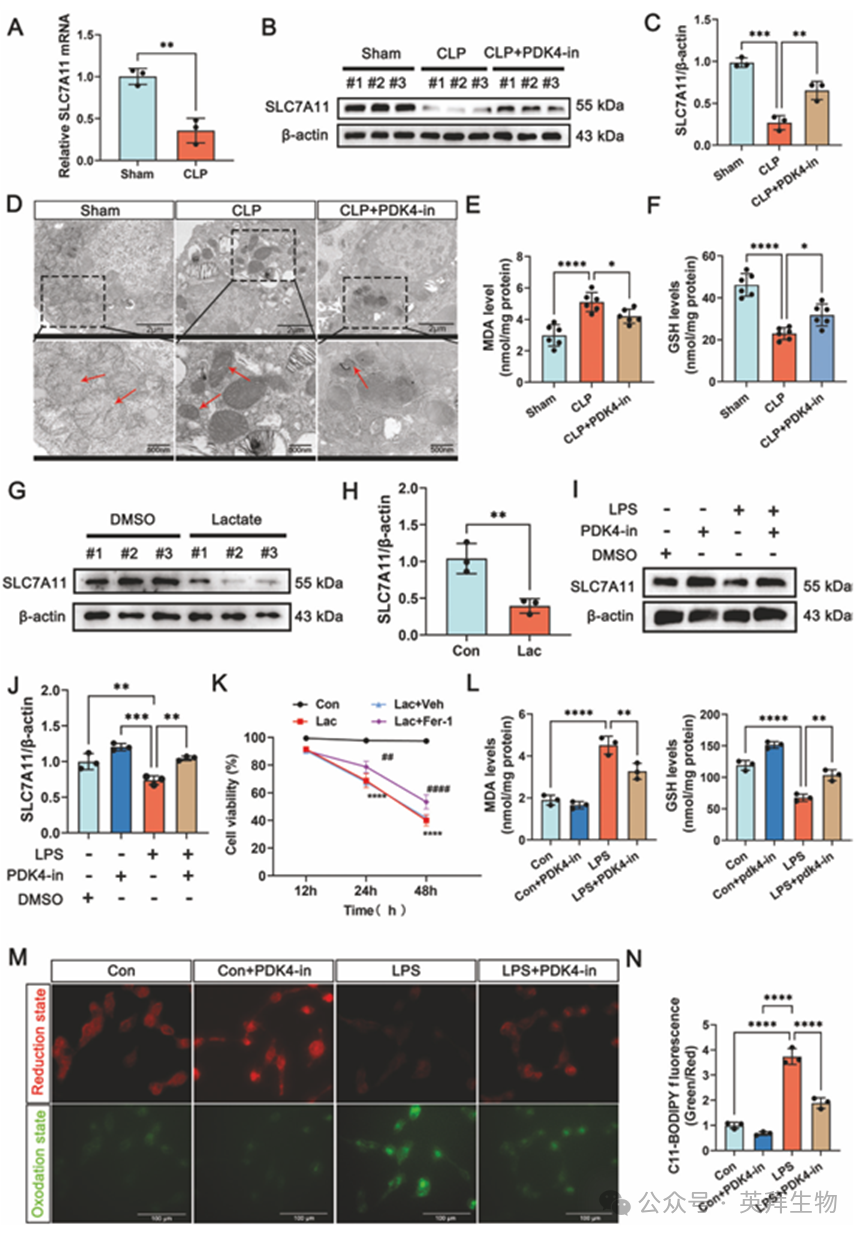
图5.乳酸通过抑制SLC7A11 加剧上皮细胞铁死亡
6.LPCAT2-K375突变通过抑制SLC7A11抑制上皮细胞铁死亡
为探究 LCAT2-K375 的乳酸化作用是否通过调节 SLC7A11 来促进铁死亡,我们利用赖氨酸到精氨酸的定点突变技术创建了 LCAT2-K375 突变体(LPCAT2-K375R)。我们将携带 LPCAT2-WT 或 LPCAT2-K375R 突变体的慢病毒转染至 BEAS-2B 细胞中。WB和Co-IP结果显示,与野生型(WT)相比,表达 LPCAT2-K375R 的 BEAS-2B 细胞中的 LCAT2-K375 水平降低了(图 6A - C)。免疫荧光染色进一步证实了这些结果(图 6D、E)。
接下来,我们评估了 LPCAT2-K375R 对细胞活力的影响。LPCAT2-K375R 的表达增强了 BEAS-2B 细胞的活力,这表明其抑制了铁死亡(图 6F)。免疫印迹分析证实,与野生型相比,LPCAT2-K375R 细胞中的 SLC7A11 表达上调(图 6G、H)。此外,这些 LPCAT2-K375R 细胞的 GSH 水平显著升高,而 MDA/ROS 水平则显著降低(图 6I - L)。总之,我们的研究结果表明,LPCAT2-K375 通过 SLC7A11 调节铁死亡。K375 突变通过增加 SLC7A11 和减少脂质过氧化来抑制铁死亡。这些结果突显了 LPCAT2-K375 在铁死亡中的关键作用及其对重症急性肺损伤的治疗潜力。
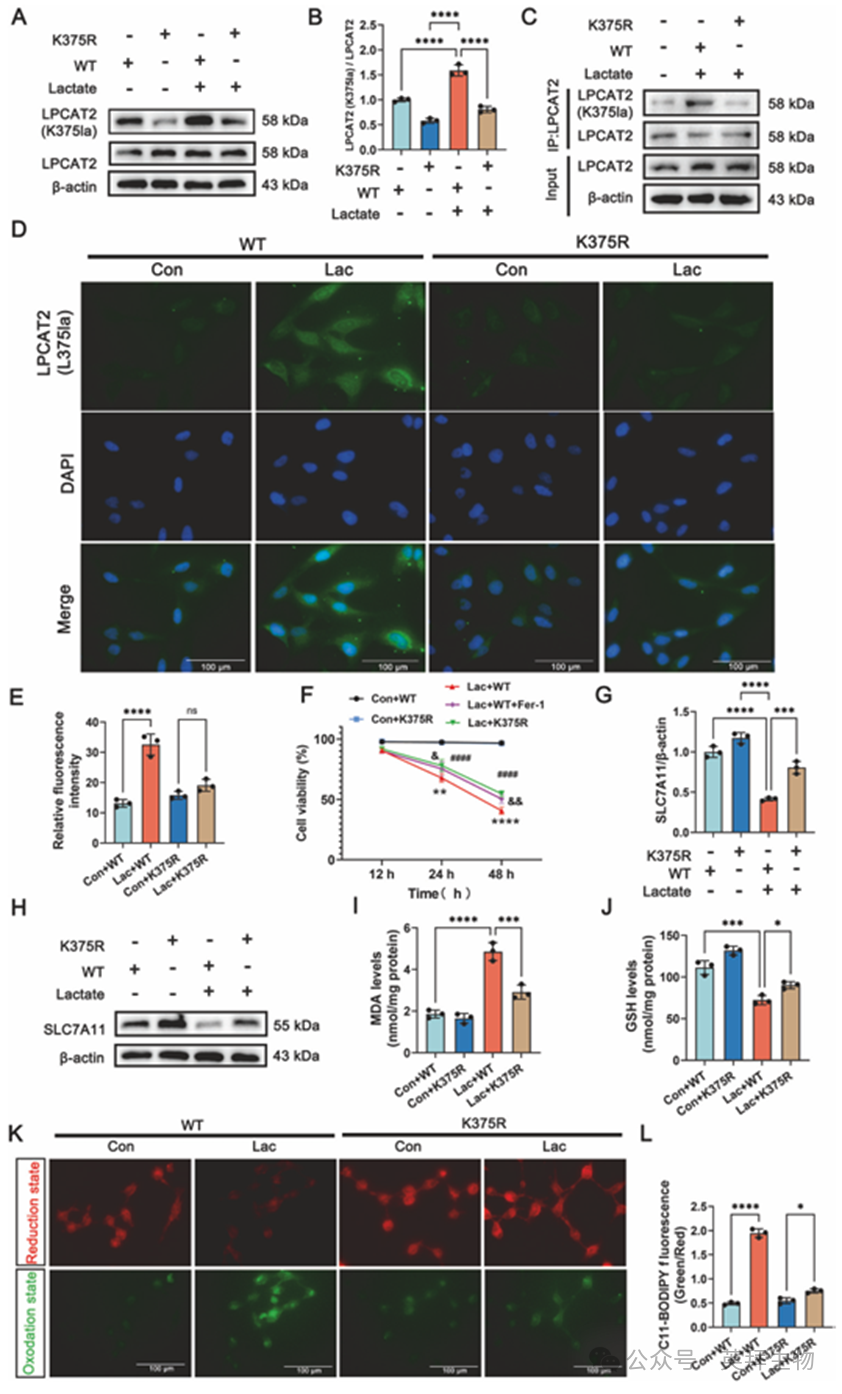
图6.LPCAT2的K375突变可减轻上皮细胞的铁死亡
7.LPCAT2-K375通过促进STAT1磷酸化和核易位转录抑制SLC7A11
为探究 LPCAT2-K375 如何调控 SLC7A11 的表达,我们进行了免疫沉淀/质谱分析,并确定 STAT1 是其相互作用伙伴(图 7A、B)。已有大量文献表明 STAT1 是 SLC7A11 表达的转录抑制因子。我们还进行了分子对接研究,以表明 LPCAT2 和 STAT1 通过氢键相互作用(图 S4B–D)。共免疫沉淀实验也表明 LPCAT2 确实能与 STAT1 结合(图 7C)。
先前的研究表明,LPCAT2 具有乙酰化酶活性。因此,我们进行了实验,并发现降低 LPCAT2 的表达会降低 STAT1 的乙酰化水平(图 7D)。有趣的是,我们还观察到,在乳酸处理条件下,STAT1 的乙酰化水平也降低了(图 7E),这表明在乳酸存在的情况下,这种活性受到了抑制。STAT1 乙酰化的减少会增加其磷酸化水平。随后,我们进行了共聚焦荧光显微镜检查和三维建模,结果显示乳酸促进了 STAT1 的磷酸化及其向细胞核的转移(图 7F)。我们还评估了 LPCAT2-K375R 细胞中的 p-STAT1 水平,结果表明 K375 突变体在高乳酸条件下显著抑制了 STAT1 的磷酸化(图 S4E、F)。CHIP 检测显示 STAT1 与 SLC7A11 启动子之间存在转录调控相互作用(图 S4G)。此前的研究报道,SLC7A11 也可受 PRMT1 的调节。然而,我们在高乳酸条件下用 PRMT1 抑制剂处理细胞后,发现 SLC7A11 的水平并未显著下调(图 3E、F)。此外,ACSL4 是铁死亡的关键调节分子之一。然而,使用 ACSL4 抑制剂 AS-252424 进行治疗未能显著降低高乳酸条件下 LPCAT2-K375R 细胞中的脂质过氧化(图 S3G、H)。这些发现表明,在高乳酸条件下,LPCAT2-STAT1-SLC7A11 轴很可能是 LPCAT2 调控通路中脂质过氧化的主要驱动因素,并且显示 LPCAT2-K375 的乳酸化通过促进 STAT1 的磷酸化和核转移来抑制 SLC7A11 的转录,揭示了 LPCAT2-K375 通过 SLC7A11 调节铁死亡的新机制。
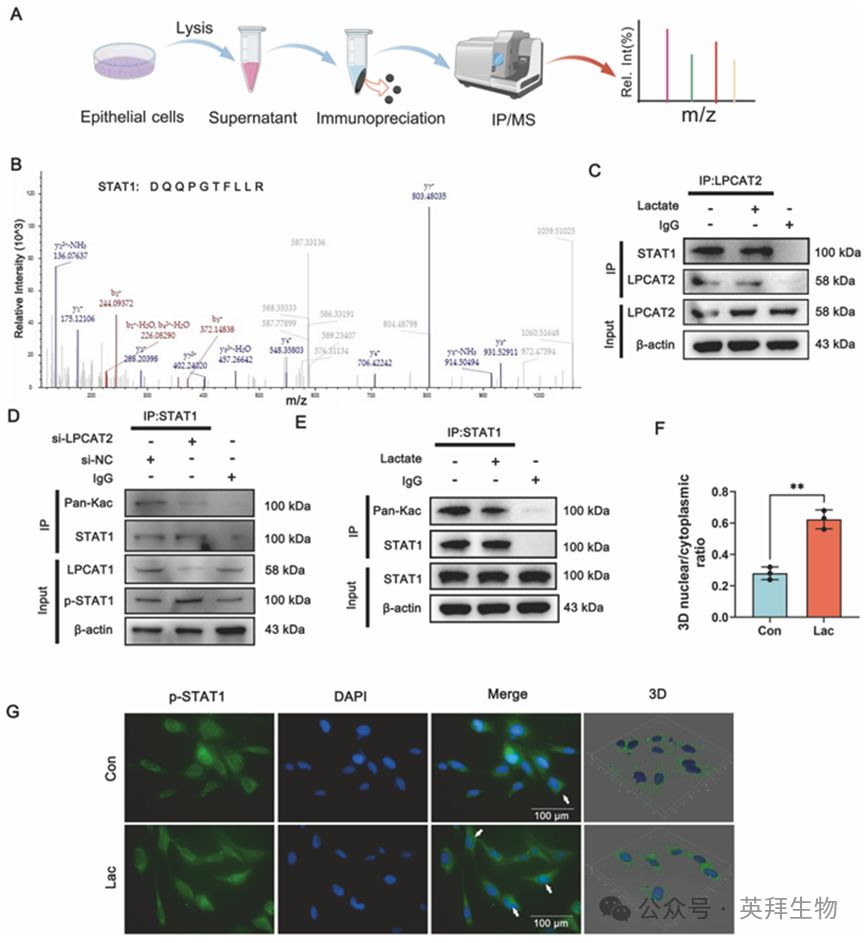
图7.LPCAT2-K375通过抑制STAT1乙酰化和促进STAT1磷酸化和核易位来抑制SLC7A11的表达
8.AARS1/HDAC9是LPCAT2-K375的乙酰转移酶/去乙酰化酶
质谱/质谱分析确定 AARS1 是负责 LPCAT2-K375 乳酸化反应的乳酸转移酶(图 8A),这一功能最近已被报道是在存在乳酸和 ATP 的条件下发挥乳酸转移酶作用的。我们还进行了分子对接研究,以表明 LPCAT2 和 AARS1 通过氢键相互作用(图 S5A–C)。共免疫沉淀实验也证实了 LPCAT2 和 AARS1 之间存在直接的相互作用(图 8B)。AARS1 在 CLP 中的上调(图 S5D–F)介导了 LPCAT2-K375 乳酸化,并且其敲低显著降低了 LPCAT2-K375 的水平(图 8C–E)。AARS1 的过表达通过 LPCAT2-K375 乳酸化上调了 SLC7A11 的表达(图 8F–H)。此外,为了检查 STAT1 的磷酸化是否调节 SLC7A11 的表达,我们过表达了 AARS1,并发现用抑制 STAT1 磷酸化的氟达拉滨处理后,SLC7A11 的水平显著增加(图 8I–K)。总之,我们的研究结果表明,AARS1 是负责 LPCAT2-K375 乳酸化的乳酸转移酶。
免疫沉淀/质谱技术进一步确认了 HDAC9 是调控 LPCAT2-K375 乳酸化的脱酰基酶(图 8L)。分子对接分析表明,LPCAT2 通过氢键与 HDAC9 相互作用(图 S6A–C)。共免疫沉淀实验证实了 LPCAT2 与 HDAC9 之间存在直接相互作用(图 8M)。我们还发现,在 CLP 组中,HDAC9 的表达水平显著降低(图 S6D–F)。
为进一步探究 HDAC9 的功能,我们降低其表达水平,结果表明,在乳酸环境中,LPCAT2-K375 的表达显著上调(图 8N–P)。此外,HDAC9 的过表达(OE)显著减少了乳酸处理细胞中 LPCAT2-K375 的乳酸化程度。这进一步支持了 HDAC9 在负向调节 LPCAT2-K375 乳酸化过程中的作用。先前的研究表明,HDAC9 可以促进 STAT1 的去乙酰化并增加其磷酸化。鉴于在 SI-ALI 中观察到的 HDAC9 表达水平的降低,我们进一步研究了在高乳酸条件下 p-STAT1 的表达情况。结果表明,磷酸化 STAT1 的水平显著增加(图 S6G,H)。这些研究结果表明,STAT1 的磷酸化主要受 LPCAT2-K375 的乳酸化状态的影响。
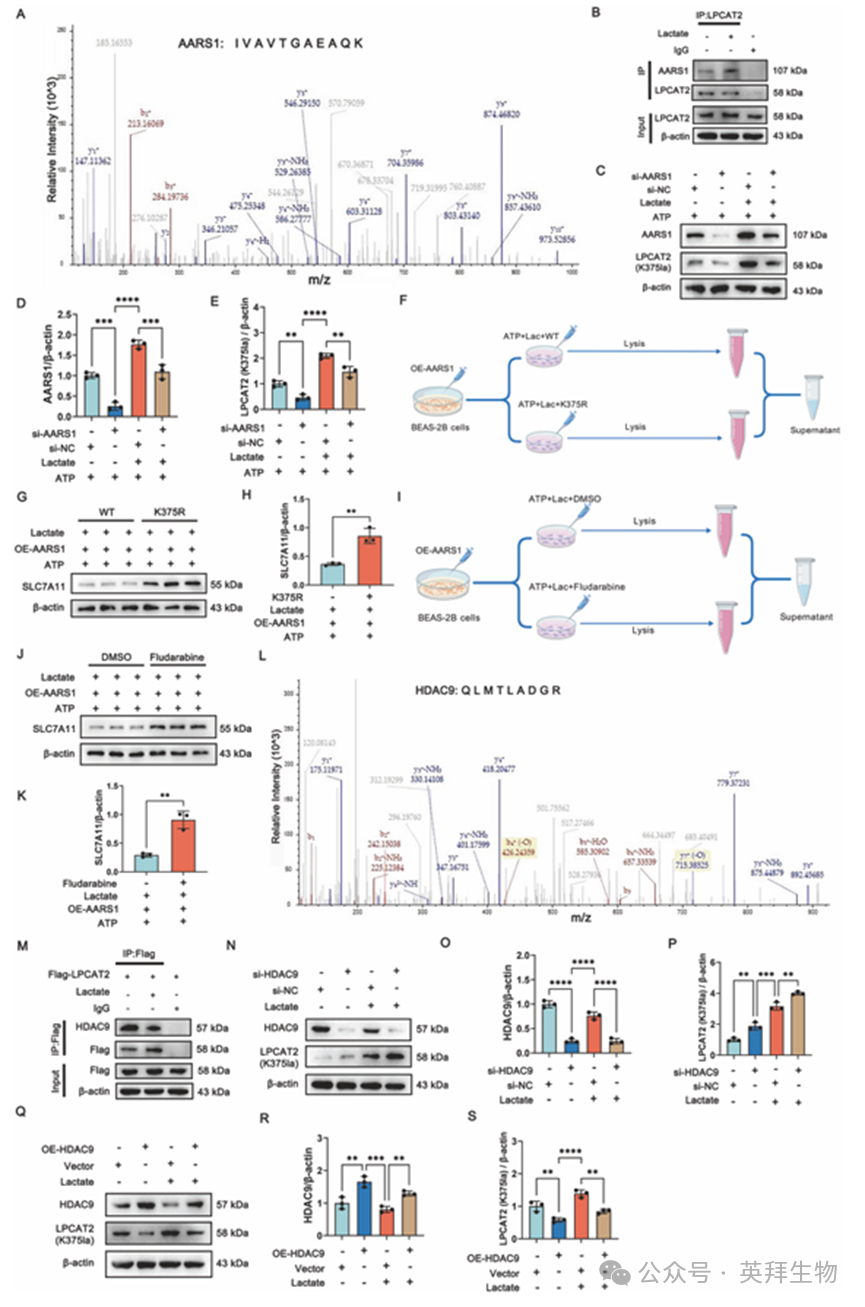
图8. AARS1/HDAC9是LPCAT2-K375的乙酰转移酶/去乙酰化酶
结论
总之,本研究强调了乳酸在重症急性肺损伤期间对上皮细胞造成损害的作用。此外,还揭示了 PDK4 抑制剂在降低重症急性肺损伤中乳酸水平方面的有效性。同时,我们还阐明了 LPCAT2 乳化作用如何调节 STAT1/SLC7A11 轴,最终导致铁死亡的过程。综合来看,我们的研究结果表明,针对 PDK4 和 LPCAT2 乳化作用的干预可能成为一种有前景的治疗策略,用于减轻脓毒症患者的肺损伤。
参考文献
Deng Y, Qiu Y, Li X, Gong T, Guo J, Liang H, Yuan Z, Hei Z, Zhang X, Liu Y. PDK4-driven lactate accumulation facilitates LPCAT2 lactylation to exacerbate sepsis-induced acute lung injury. Cell Death Differ. 2026 Mar;33(3):557-573. doi: 10.1038/s41418-025-01585-6. Epub 2025 Oct 7. PMID: 41057687; PMCID: PMC13035903.