






肿瘤基质“糖变”加剧免疫抑制,去糖基化重塑TNBC微环境
肿瘤ECM(ECM)越来越被认为是肿瘤免疫抑制和治疗抵抗的关键驱动因素。然而,目前对造成这种免疫抑制环境的具体ECM成分和机制仍知之甚少,阻碍了新疗法的开发。在这里,我们使用三阴性乳腺癌(TNBC),一种侵袭性和耐药的亚型,全面的多组学分析来研究这个问题。我们发现TNBC中的ECM免疫调节是由ECM蛋白的翻译后糖修饰介导的。利用去细胞的人TNBC样本,我们发现靶向酶去除这些ECM聚糖可以改变肿瘤免疫微环境。这种修饰将肿瘤相关髓系细胞重新编程为免疫调节表型,并改善T细胞浸润。值得注意的是,ECM脱氨酰化改变了T细胞上的选择素和选择素配体程序,这与运输和肿瘤内通路的改善一致。与此同时,巨噬细胞-T细胞的相互作用被重塑,导致T细胞耗竭减少。我们的研究发现,ECM聚糖修饰是先天和适应性TNBC免疫微环境的关键调节因子。他们认为,靶向ECM糖基化可能提供增强这种侵袭性乳腺癌亚型抗肿瘤免疫的潜在策略。该研究于2026年6月发表在《Nature Communications》,IF:18.1。
技术路线
主要研究结果:
1. TNBC的多层特征揭示了ECM失调
之前,我们发现了一个与肿瘤相关的ECM标签,称为基质指数(MI),它是高级别浆液性卵巢癌(HGSOC)预后不良的指标,并且在13种主要癌症类型中与免疫抑制性免疫细胞类型和预后价值增加相关,在TNBC10中最显著。在本研究中,我们进一步探索了MI高、预后差的TNBC组织中的失调通路(图1a,补充图1a),发现这些组织中富集了与N-连锁糖基化和适应性免疫抑制相关的基因(图1b,补充图1b),支持以下假设:在TNBC中,ECM蛋白上显示的聚糖使肿瘤ECM具有抗肿瘤免疫。
我们使用RNA-seq(补充数据1,补充图1c -e)、ECM富集的蛋白质组学(补充数据2)、n -连锁糖组学(补充数据3)和空间免疫细胞谱(图1c)分析了32例TNBC患者的原发肿瘤组织和12例患者匹配的组织病理学正常的邻近/周围组织(“邻近”)。在基因(图1d)和蛋白水平(图1f)评估癌旁组织和肿瘤组织之间ECM的变化。我们确定了245个显著失调的ECM基因(补充数据4)和31个ECM蛋白(补充数据5),两个数据集之间有16个重叠(图1g)。进一步将ECM成分分类为核心基质体蛋白(蛋白聚糖、ECM糖蛋白和胶原)和基质体相关蛋白(分泌因子、ECM相关蛋白和ECM调节因子),发现与邻近组织相比,肿瘤组织中糖基化的ECM蛋白(“ECM糖蛋白”)增加,这与MI呈正相关(图1e)。在失调的分子中,基因和蛋白质数据集之间的大部分重叠被归类为ECM糖蛋白(图1h),包括纤维连接蛋白(FN1)和基质重塑相关蛋白5 (MXRA5),我们之前在HGSOC18中将其与免疫抑制巨噬细胞表型关联。这些分析描述了TNBC中ECM成分的变化,其中ECM糖蛋白是病变ECM的突出特征。
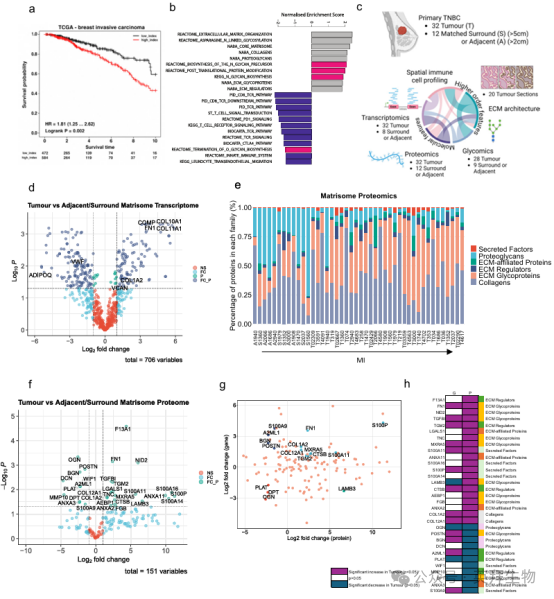
图1. TNBC的多层特征揭示了ECM的失调
2. 异常的ECM翻译后修饰与免疫细胞浸润密切相关
在发现ECM糖蛋白是肿瘤ECM的一个显著特征的基础上,我们想进一步确定与之相关的聚糖。我们首先研究了TNBC患者的癌旁组织和肿瘤组织之间,在转录水平上糖基化生物合成途径的变化,发现糖基化生物合成基因显著失调(图2a)。将这些基因注释为聚糖生物合成家族表明,肿瘤和邻近组织分为两个不同的簇,其中肿瘤组织显示N -连接和O-连接的聚糖生物合成过程上调,包括聚焦化、聚- LacNAc合成和唾液酸化(图2b)。GlycoMaple是一种经过验证的基于基因表达估计聚糖结构的工具,该工具证实了这些发现,并预测肿瘤组织中N -连接聚糖唾液化、岩藻糖基化和poly-LacNAc结构显著增加(图2c)。我们通过将患者组织分为细胞、膜和ECM区室,并对ECM富集的部分进行N-连接糖组学,证实了这些N-糖聚糖结构在ECM中的存在。这证实了如在转录组水平预测的那样,ECM上的唾液酸lewis(补充图2a-c)、聚焦化(补充图2d-f)、poly-LacNAc(补充图2g-i)和唾液酸化(图2d-f)增加。此外,我们观察到肿瘤组织向复杂性较低的高甘露糖N -聚糖转变,这与之前的文献一致(图2d, e)19。在细胞膜部分中观察到类似的变化(补充图3)。
对ECM(补充图4)和细胞膜(补充图5)上的oO-连接聚糖进行的分析显示,Sialyl-Tn抗原(STn)(补充图4c)和唾液酸化聚糖(补充图4e, f)增加。然而,在我们的样本中,o -聚糖的检测有限(补充图4a)。使用凝集素免疫荧光染色,我们证实,与邻近组织相比,肿瘤组织ECM上的唾液酸化、聚lacnac、高甘露糖N -聚糖和O-聚糖显著增加(补充图6)。将xCell(一种分析肿瘤免疫微环境的计算机工具)应用于与患者匹配的N -糖链数据(补充图7),我们发现高甘露糖和高唾液酸化的N -糖链(二唾液酸化、三唾液酸化和四唾液酸化)与T细胞、树突状细胞和B细胞标签之间存在中度正相关,提示ECM糖与TME中的适应性免疫浸润之间存在关系(图2g, h)。
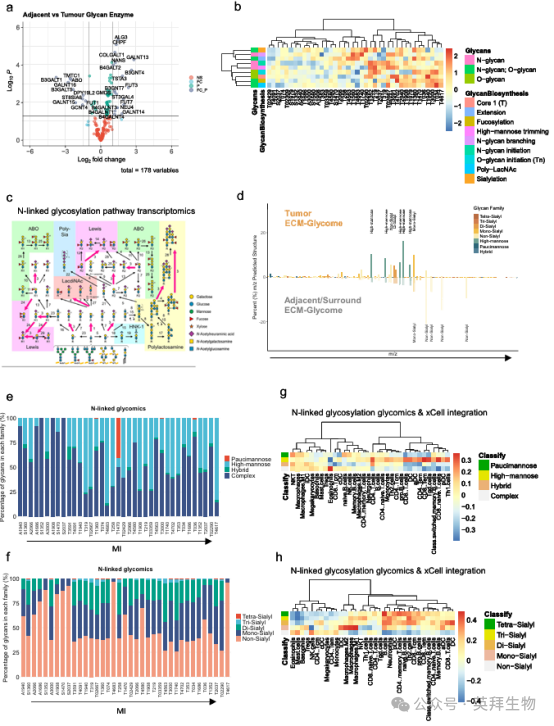
图2. TNBC的多层表征显示ECM翻译后聚糖的改变与免疫细胞浸润显著相关
3. TNBC TME富含免疫排斥表型,与T细胞定位相关的N-连锁糖基化和唾液酸化
批量RNA-seq和ECM蛋白质组学缺乏空间分辨率,因此我们使用与我们的ECM分析匹配的20例FFPE患者组织的免疫组化方法,对CD8+ T细胞进行了空间免疫细胞谱分析(图3a)。正如我们之前所描述的,我们将组织分层为3类:27%的“炎症”肿瘤,CD8+ T细胞浸润到肿瘤核心,44%的“沙漠”肿瘤,总体CD8+ T细胞浸润较低,27%的“排除”肿瘤,免疫细胞被困在肿瘤巢外(图3b-c)。我们根据炎症和排除的肿瘤表型对数据集进行分层,并将这一分析与患者匹配的N -连锁糖组学数据整合,结果显示,与炎症组织相比,排除的组织表现出显著升高的唾液酸化(图3d-i)。对RNA-seq或蛋白质组学数据进行的分层表明,在炎症表型和排除表型之间,在转录或蛋白质水平,基质体基因或蛋白质无显著差异(补充图8a, b)。
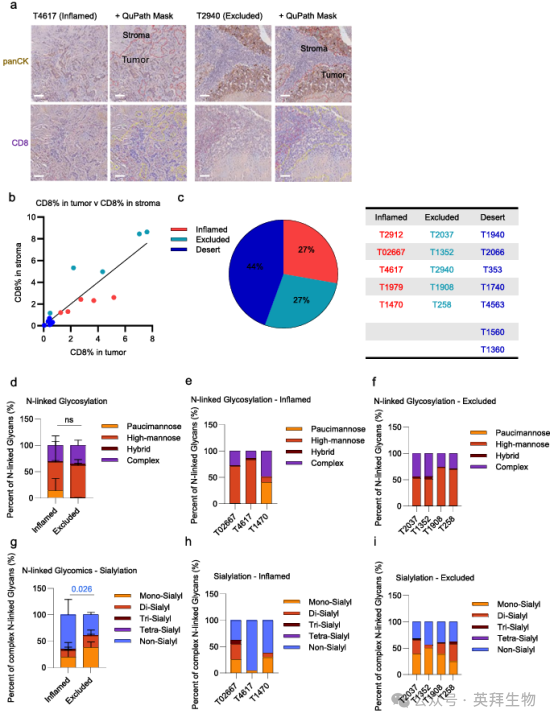
图3. TNBC TME富含免疫排斥表型,与T细胞定位相关的N-连锁糖基化和唾液酸化
4. 脱细胞的TNBC组织表现出免疫排斥的TME,通过修饰ECM聚糖可逆转
为研究基质糖基化对体外TME的影响,我们根据我们之前发表的方案,开发了TNBC的肿瘤ECM脱细胞组织模型。简单地说,用振动刀将组织切片成350 µm切片,然后进行一系列洗涤,裂解细胞,提取脂质,降解残留核酸(图4a)。通过评估细胞去除情况来确认脱细胞(图4b),之后发现ECM结构(图4c和补充图10)、机械性能(图4d)、糖基化模式(补充图10)和成分(图4e和补充图9)在后处理过程中保持完整。
为验证脱细胞组织用于细胞培养和测试ECM糖基化的免疫功能,我们首先评估了模型中的癌细胞活力。TNBC细胞株BT20和HCC38在癌旁组织或肿瘤组织脱细胞基质上培养7 d。值得注意的是,与生长在胶原基质上的细胞相比,TNBC细胞在整个培养期间保持了较高的活力(图4f, g)。
在建立了适合研究ECM糖基化功能的模型后,我们现在将注意力转向研究免疫细胞的活性。我们之前在多层组学分析中建立了ECM糖基化和T细胞浸润之间的关系(图2f-h, 3),因此我们使用前文所述的首先用TNBC细胞进行再细胞化的脱细胞组织模型,询问ECM糖基化是否直接参与了CD8+ T细胞浸润。我们开发了一种活细胞成像分析方法,用于监测原代T细胞或靶向TNBC的CAR T细胞在ECM内的移动和定位(图5a,补充图11)。我们使用CAR T细胞作为天然肿瘤反应性T细胞群的替代指标。用HCC38再细胞化的组织用从健康供者外周血单个核细胞(PBMCs)中分离的原代T细胞培养,而用BT20再细胞化的组织用CAR T细胞培养。首先,我们观察到,在未使用TNBC细胞进行再细胞化的脱细胞组织中,CAR t细胞浸润极少(补充图12a)。在这些组织中,CAR t细胞的运动速度比再细胞组织慢(补充图12b),这表明癌细胞的存在刺激了CAR T细胞的浸润。比较肿瘤来源的ECM和邻近组织之间的原代T细胞或CAR T细胞浸润,我们发现T细胞和CAR T细胞浸润在肿瘤来源的ECM中显著减少(图5b, c,补充图13b, c)。此外,与邻近组织相比,在肿瘤ECM中穿透肿瘤巢的T细胞或CAR T细胞较少(距离癌细胞≤5µm)(图5d、补充图13d和补充图14),类似于免疫排斥表型。在肿瘤和癌旁组织中,T细胞或CAR T细胞优先聚集在间质中,这与人卵巢和肺肿瘤新鲜切片的实时显微镜下记录的常驻CD8+ T细胞的行为一致(图5d,补充图13e, f)。
T细胞运动分析显示,与肿瘤组织相比,位于HCC38癌细胞附近(<5µm)的T细胞在邻近组织中的速度和位移增加(图5e, f)。此外,在肿瘤ECM中,与周围ECM相比,位于肿瘤胰岛附近(HCC38)的T细胞表现出速度和位移的下降(图5g, h)。这些观察结果与之前的报道一致,即密集的细胞外基质结构限制了T细胞的迁移,并限制了它们穿透和迁移到肿瘤胰岛。
正如我们之前在多组学分析中观察到的T细胞和N -糖基化,尤其是唾液酸化之间的关系(图2h,图3),我们想测试用酶去除N -糖基或唾液酸是否显著增强T细胞浸润。我们首先通过PNGase F处理脱细胞组织来切割N-聚糖,或者通过产气荚膜梭菌的神经氨酸酶处理脱细胞组织来去除N-和o -连接的聚糖上的末端唾液酸,从而确认该模型对酶修饰敏感,这一点通过凝集素染色得到了证实(补充图15)。
酶解法去除N -聚糖或唾液酸显著增强了T细胞和CAR T细胞在肿瘤ECM中的浸润(图5i, j,补充图13i, j和补充图16),并增加了T细胞或CAR T细胞穿透肿瘤巢的比例(图5k, l,补充图13k和补充图16)。有趣的是,与未处理的肿瘤组织相比,当靠近癌细胞时,PNGase F或神经氨酸酶处理组织中的T细胞表现出更大的速度和位移(<5µm),这表明T细胞更容易通过酶处理组织中的ECM到达肿瘤胰岛(图5m, n)。
然而,对与CAR T细胞共培养的BT20进行的相同分析表明,与周围ECM中的CAR T细胞相比,位于肿瘤胰岛附近的CAR T细胞表现出更快的速度和位移(补充图13g, h)。这一观察结果与先前的报道一致,即肿瘤胰岛可作为T细胞迁移增强的区域,并且也表明在该模型中,癌细胞对T细胞迁移的特异性效应也很活跃。在BT20和CAR T细胞在邻近组织共培养时未观察到这一现象,提示肿瘤ECM在调节CAR t细胞行为中的特定作用(补充图14d, e)。此外,在PNGase F或神经氨酸酶处理的组织中,CAR T细胞在ECM中表现出更大的速度(>5µm),表明CAR T细胞更容易在酶处理的组织中通过ECM(补充图13l, m)。
总之,我们的结果表明,酶促治疗肿瘤ECM可以逆转免疫排斥表型。
为探索糖调节T细胞浸润的机制,我们聚焦于唾液酸- siglec轴,这是一个已知的免疫抑制通路,可能与肿瘤细胞外基质上的唾液酸化相关。我们对接种在肿瘤组织或经神经氨酸酶治疗的肿瘤组织上的CAR T细胞的Siglec受体表达进行了分析,结果显示约5%的细胞毒性CD8 + T细胞表达Siglec-7, 10%表达Siglec-10,两种情况无显著差异(补充图17,18)。虽然T细胞上的Siglec-7和Siglec-10表达水平在ECM去唾液酸化后无变化,但这并不排除siglec介导的信号传导发挥作用,因为神经氨酸酶治疗主要改变配体的可用性,而不是受体的表达。我们还观察到,PNGase F或神经氨酸酶酶促治疗改变了组织结构,特别是增加了ECM纤维的排列,这可能会增强CAR T细胞或T细胞对肿瘤的接触(补充图19、20)。综上所述,这些发现表明,修饰ECM聚糖可以将免疫排斥的TME重新编程为免疫炎症表型。值得注意的是,从ECM中去除聚糖会导致纤维结构的重组。虽然这一现象在本文中没有进一步探讨,但它可能源于ECM纤维之间改变的聚糖-聚糖或聚糖-蛋白相互作用,和/或来自通常通过聚糖介导的结合而保留的ECM相关因子的释放。这种结构变化可能通过共同影响糖依赖的信号传导、基质组织和组织的物理性质来影响T细胞浸润。
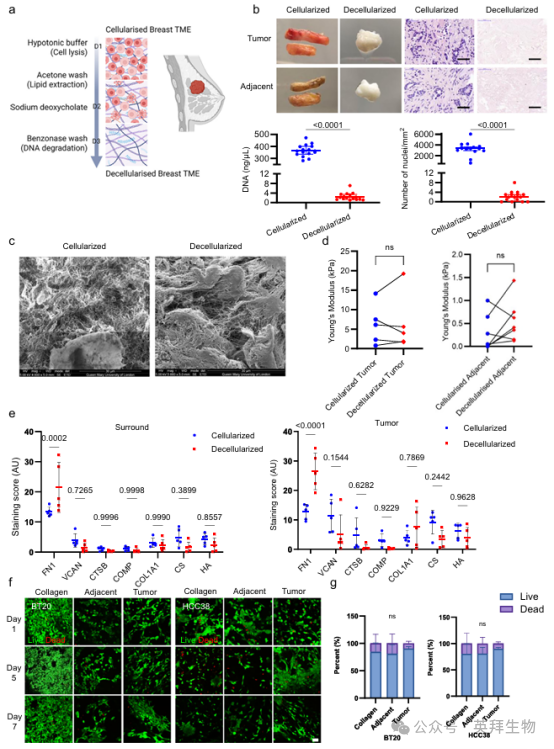
图4. TNBC脱细胞组织模型的开发,以测试针对失调肿瘤TNBC ECM的新疗法
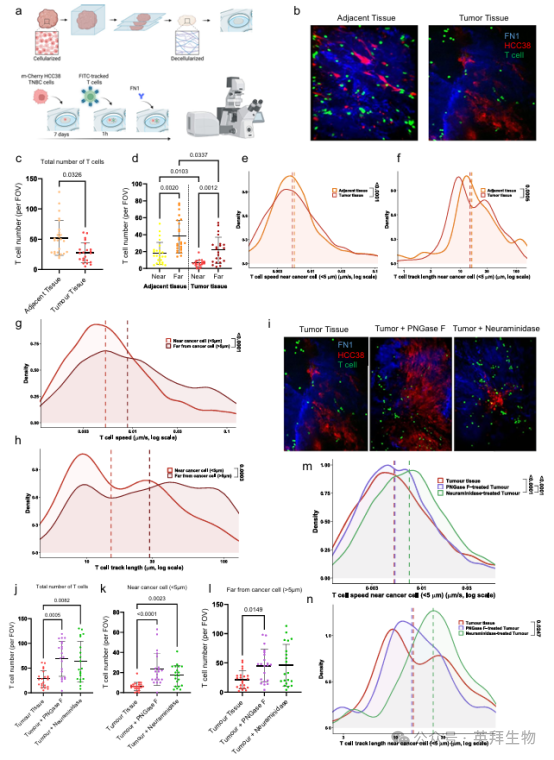
图5. 脱细胞的TNBC组织维持免疫排斥的TME,通过聚糖修饰转化为免疫炎症表型
5. TNBC TME诱导由ECM聚糖介导的免疫调节巨噬细胞群
成功的适应性抗肿瘤免疫应答在很大程度上依赖于固有免疫细胞的支持,尤其是巨噬细胞通常是支持T细胞活性抑制肿瘤所必需的。我们之前观察到,肿瘤ECM可以直接影响HGSOC18中肿瘤相关巨噬细胞表型的行为。为了测试这种基质培养的巨噬细胞(MAM)是否也是TNBC的特征,我们使用了我们的脱细胞组织模型。将健康供者PBMCs中分离出的单核细胞在脱细胞的肿瘤组织和癌旁组织上培养,14 d后收集巨噬细胞(图6a,补充图21)。我们通过对细胞表面标志物(包括CD11b、CD209、CD206、CD163、CD86、HLA-DR、Siglec-1和Siglec-9)进行的流式细胞术分析评估了巨噬细胞表型转化(补充图21b)。
我们观察到肿瘤组织上的CD11b+ MAMs显著少于邻近组织(补充图22a)。鉴于CD11b参与调节髓系细胞极化和功能状态,这一转变提示,肿瘤ECM暴露可能与巨噬细胞分化或免疫调节程序的改变相关。
我们之前在多组学分析中观察到巨噬细胞和唾液酸化之间的关系(图2h),并且HGSOC中MAMs的转录组谱与已发表的MUC1刺激的高唾液酸化巨噬细胞数据集相关。为测试ECM上的唾液酸化是否影响TNBC中的MAM表型,我们使用神经氨酸酶处理肿瘤ECM,该酶显著增加了CD11b +、CD163+和CD209+巨噬细胞的比例(图6b)。这种效应在神经氨酸酶处理的邻近ECM中没有观察到,这表明只有唾液酸在肿瘤ECM中显示为巨噬细胞表型的调节剂。免疫调节标志物(CD163、CD206、CD209)的组合流式分析显示,在经神经氨酸酶处理的肿瘤组织中,共表达两种免疫调节标志物的巨噬细胞比例显著增加(图6c、d),提示ECM唾液酸在TME中巨噬细胞表型的免疫调节变化中发挥作用。对抗原呈递标志物(CD86、HLA-DR)的组合分析显示无显著变化(图6e)。在所有情况下,大多数巨噬细胞表达Siglec-1和/或Siglec-9,这凸显了唾液酸- siglec相互作用在巨噬细胞表型调节中的潜在机制(补充图22b)。
从整个组织中,对组织唾液化水平的分析表明,组织唾液化水平与CD11b+巨噬细胞分化呈显著负相关。肿瘤ECM的高唾液酸化与CD45+ CD11b+巨噬细胞减少相关,这与我们的发现一致,即神经氨酸酶介导的去唾液酸化增加了CD45+ CD11b+巨噬细胞群(补充图22c, d)。这些结果强调了肿瘤ECM唾液化在巨噬细胞表型形成中的作用,并提示TME中唾液酸水平和免疫调节之间的功能联系。
正如我们之前所表明的,在肿瘤组织中培养的MAMs可影响T细胞增殖和耗竭标志物,我们试图确定TNBC中的MAMs是否调节T细胞功能。我们进一步测试了来自癌细胞(CC)条件培养基的可溶性因子是否影响MAMs和T细胞的相互作用以及下游T细胞应答(图7a,补充图23)。供者匹配的T细胞与在肿瘤或邻近组织上培养的MAMs(有或没有神经氨酸酶处理),以及在有或没有CC培养基的情况下进行培养,以确定这些巨噬细胞是否直接调节T细胞表型。活化标志物(CD137和ICOS)的组合分析显示,与肿瘤组织上MAMs共培养的T细胞相比,肿瘤组织上MAMs的活化显著增加。此外,在癌旁组织中加入癌细胞可溶性因子降低了与MAMs共培养的T细胞的活化,而在肿瘤组织中,癌细胞培养基对T细胞活化的影响减弱(图7b)。这表明,肿瘤ECM对T细胞活化具有主导影响,而不考虑癌细胞衍生的可溶性因子的存在。此外,与癌旁组织相比,与肿瘤组织MAMs共培养的T细胞中,三种耗竭标志物(PD1, LAG3和TIM3)的组合表达显著增加(图7c, d,补充图23b)。有趣的是,神经氨酸酶治疗显著降低了肿瘤组织中与MAMs共培养的T细胞上的耗竭标志物,而仅导致邻近组织中MAMs培养的T细胞上的耗竭标志物轻微降低(图7c, d,补充图23b)。这一结果表明,高唾液酸化的肿瘤ECM在指导这种免疫抑制性巨噬细胞- T细胞表型中的作用。重要的是,在任何条件下,癌细胞可溶性因子对耗竭标志物的表达没有显著影响。有趣的是,在经神经氨酸酶处理的去细胞肿瘤组织上单独培养的T细胞表现出最小的耗竭标志物表达降低(基于两种组合标志物),而在去细胞的邻近组织中未观察到这一情况。然而,单独ECM培养的T细胞整体耗竭水平高于与MAMs共培养的T细胞。这表明,对T细胞表型的主要影响来自与MAMs的相互作用,而不是来自ECM唾液化对T细胞的直接作用(补充图24a, b)。
为进一步探索ECM在巨噬细胞- T细胞交叉对话中的免疫调节功能,我们对肿瘤和邻近组织的脱细胞模型中T细胞和巨噬细胞共培养的上清液进行了细胞因子谱分析。细胞因子谱分析显示,在肿瘤组织上的MAMs和T细胞共培养的上清液中,IFN-γ和TNF-α减少,表明肿瘤ECM影响免疫细胞的交叉对话,这可以通过减少免疫炎性细胞因子释放来证明(图7e和补充图24c)。有趣的是,对邻近组织和肿瘤组织进行神经氨酸酶治疗减少了抗肿瘤细胞因子(如IL-6、IL-10、IL-17A和IL-23)的释放(图7e和补充图24c),表明唾液酸去除减少了促肿瘤细胞因子的产生,抑制了免疫抑制,这与在耗竭表型中观察到的减少相关(图7c, d)。
最后,我们试图进一步阐明ECM脱唾液酸增强T细胞浸润的机制。虽然我们没有注意到Siglec表达的变化(补充图17,18),但我们想知道去除唾液酸是否会影响T细胞上选择素或选择素配体的表达。在我们的模型中,皮肤淋巴细胞抗原(CLA)、CD62L (l -选择素)和CD162 (p -选择素糖蛋白配体-1,PSGL-1)单独在T细胞上高表达(补充图24d, e)。有趣的是,与癌旁组织相比,肿瘤组织中MAMs培养的T细胞显著增加了CD62L和CD162(图7f,补充图23c)。这提示肿瘤ECM影响CD62L和CD162高表型,而已知CD62L和CD162高表型会减弱T细胞的细胞毒性作用,并表明其浸润能力较差。经神经氨酸酶治疗后,肿瘤组织中CD62L和CD162的表达显著降低,表明细胞毒性表型改善,细胞耗竭减少,肿瘤组织浸润更好。
总之,我们的结果表明,ECM去唾液酸化增强了肿瘤巢内的T细胞浸润和运动,重塑了巨噬细胞的免疫调节程序,并通过MAMs-T细胞相互作用减少了与前肿瘤细胞因子释放减少相关的T细胞耗竭。与此同时,ECM脱巯基化还可调节T细胞上选择素和选择素配体的表达,从而促进更强的细胞毒性和浸润能力表型,共同形成更利于免疫的肿瘤微环境。
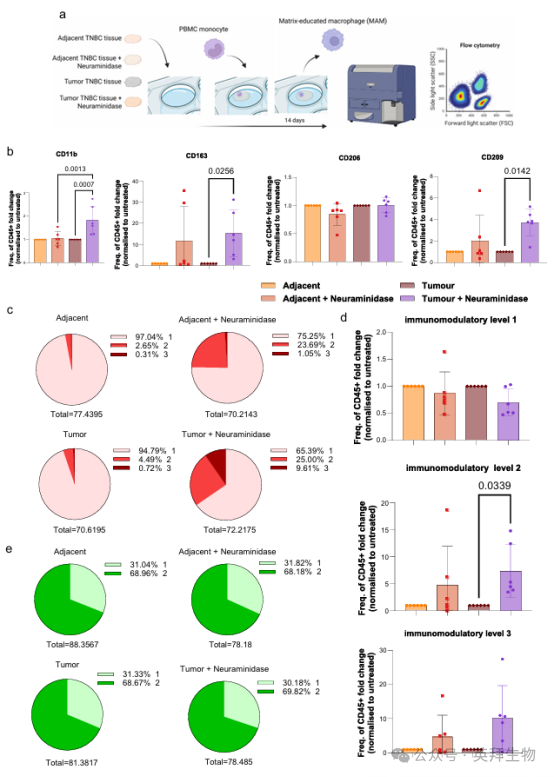
图6. TNBC TME诱导由ECM聚糖介导的肿瘤相关巨噬细胞群
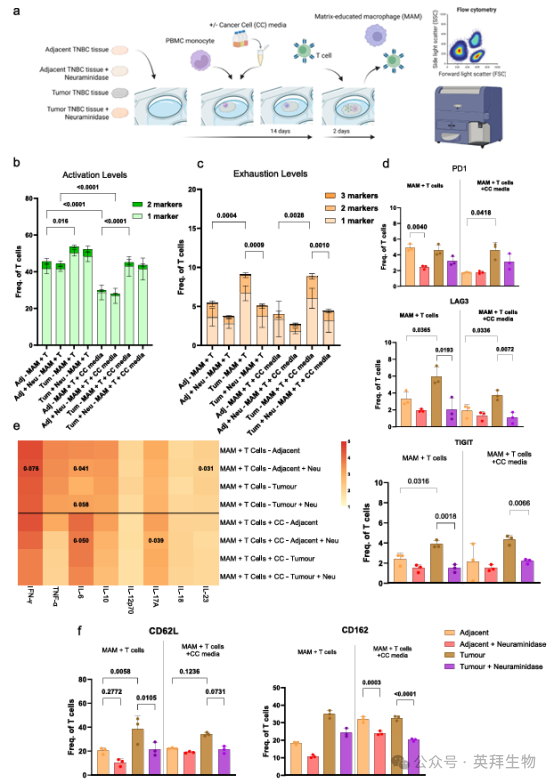
图7. MAMs以聚糖依赖的方式影响T细胞表型
结论
综上所述,我们的研究结果揭示了异常的ECM糖基化是TNBC免疫排斥的一个关键调节因子,虽然通过直接相互作用或通过改变细胞类型之间的交互作用,但以不同的方式调节巨噬细胞和T细胞的免疫。旨在编辑ECM上糖基化模式的疗法可以改善TME中免疫细胞的位置和表型,使肿瘤对免疫细胞介导的破坏更敏感。促进这一过程所需的相对较小的ECM变化可能有助于限制脱靶或不必要的效应,这些效应在靶向ECM生物合成的方法中更有可能发生。最后,由于可以阻断酶活性的小分子药物、可以替代天然糖同时改变其免疫生物学的拟糖药物或对显示的糖结构的酶降解,因此糖的生物合成和提呈可以被打断,因此糖应被视为有吸引力的治疗设计靶点。这些发现提示,为充分了解TME如何调节免疫,并开启以ECM为重点的治疗新时代,我们必须超越转化,而着眼于转化后的肿瘤微环境。
参考文献
Tarantola L, Tyler EJ, Liu Y, Maniati E, Thornton KA, Martín-Otal C, Hanna D, Kumar R, Gauthier V, Hirani P, Burger Ramos M, Roth NJ, Bragg J, Puttock EH, McDermott J, Rajeeve V, Cutillas P, Maiques O, Soulier A, Correa de Sampaio P, Jones LJ, Davies DM, Maher J, Haslam SM, Läubli H, Pearce OMT. Glycosylated extracellular matrix drives immune suppression by modulating macrophage-T cell crosstalk in triple-negative breast cancer. Nat Commun. 2026 Jun 16;17(1):5008. doi: 10.1038/s41467-026-73467-5. PMID: 42303597; PMCID: PMC13273098.