






肥胖病研究新火花:m6A甲基化可调控自噬和脂肪形成
肥胖,一类由脂肪细胞的体积和数量增加所引起的脂肪组织的异常或过度积累的疾病,已然成为人们关注的焦点,提高对于脂肪生成分子机制的理解具有重要的科学意义。研究人员已提出许多调节脂肪细胞分化的机制,包括细胞外信号,转录级联和表观基因修饰等。近日,这篇文章提出了肥胖研究的新奇思路,甲基化和自噬之间能碰撞出什么火花呢,我们来一睹为快吧。

文章思路:

1.FTO促进自噬并进一步增强脂肪形成
探讨FTO在自噬中的作用:3T3-L1细胞中FTO敲低显著降低LC3-II:I比值和增加SQSTM1水平,表明没有稳态自噬体形成。相反,过度表达HA标记的FTO显着提高LC3-II:I比值和减弱SQSTM1表达,表明FTO与自噬之间存在正相关关系。FTO的沉默减少自噬体的数量, 减轻自噬的激活。为确定自噬是否受FTO过表达影响, 3-MA或巴弗洛霉素A1处理实验进一步证实FTO促进自噬的激活。
检测FTO是否通过自噬影响脂肪形成: 3-MA和Baf A1处理可以逆转FTO过表达细胞中增强的自噬、脂肪生成和甘油三酯的积累。脂肪细胞标记基因,包括Pparg、Fabp4和Cebpa在FTO过表达细胞中显着升高,3-MA或Baf A1处理可以下调到正常水平。这些结果表明FTO通过促进自噬增强脂肪细胞分化。
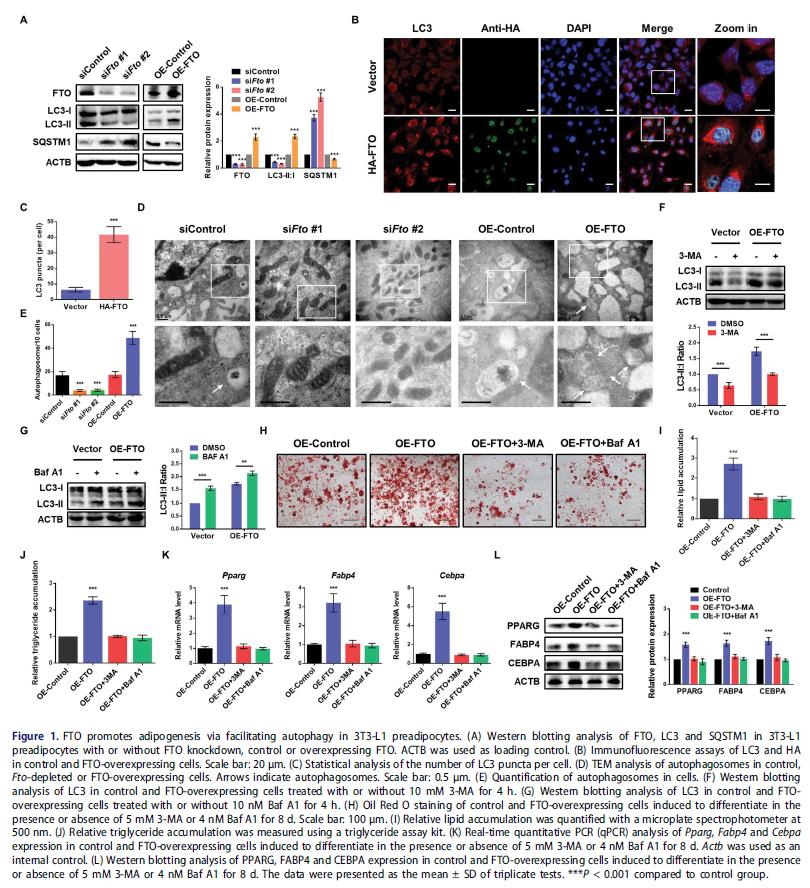
2. 敲低FTO可降低ATG5和ATG5的表达,但没有降低ULK1的表达
鉴定FTO在自噬中的潜在靶基因:检测FTO敲低后自噬相关基因的表达,Atg5和Atg7水平均减弱,而ULK1等自噬相关蛋白,如ATG12和ATG16L1,没有显着改变。检查脂肪细胞分化过程中ATG5和ATG7的表达谱,发现它们都在早期有所增加,之后都减少,这与FTO的表达模式相似。这些表明FTO的靶基因可能是Atg5和Atg7,而不是Ulk1。
检测ATG5和ATG7对自噬和脂肪生成的影响:3T3-L1脂肪前体细胞敲低Atg5和Atg7, LC3II:I比值降低, FTO蛋白表达没变化。FTO,ATG5和ATG7独自敲低都会抑制脂肪生成和甘油三酯积累,脂肪细胞标记基因水平下调。
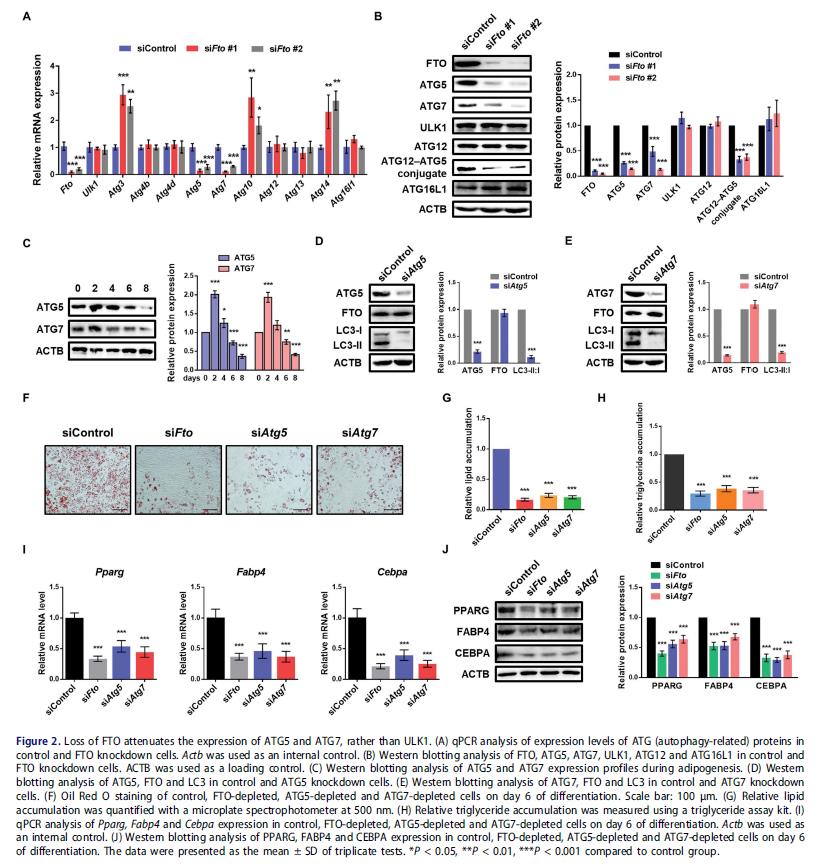
3.FTO靶向 ATG5和ATG7并影响自噬和脂肪形成
FTO过表达增加Atg5和Atg7的mRNA水平,沉默ATG5可逆转FTO过表达3T3-L1细胞中上调的LC3-II:I比例, ATG7效果一样,表明FTO通过调节ATG5和ATG7的表达影响自噬。前人研究发现ATG5和ATG7敲低抑制自噬并减少CEBPB水平,抑制PPARG和CEBPA表达损害脂肪形成分化,提示FTO通过Atg5和Atg7-Cebpb信号调节脂肪生成。FTO强表达促进CEBPB蛋白水平,而ATG5和ATG7的敲低逆转CEBPB的表达(图3D)。ATG5和ATG7的敲低恢复了FTO过表达3T3-L1细胞的脂肪生成 和甘油三酯积累, FTO过表达细胞中Pparg,Fabp4和Cebpa的mRNA和蛋白水平也被逆转。FTO通过调节Atg5和Atg7-Cebpb信号轴促进脂肪生成。
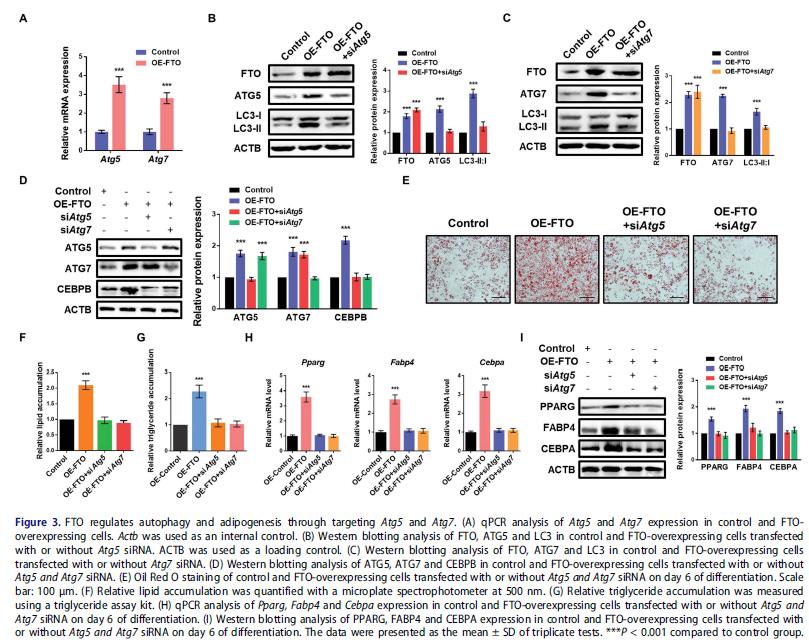
4.FTO以 m6A依赖方式调节ATG5和ATG7的表达
构建野生型(FTO-WT)和突变的FTOR96Q(FTO-MUT)质粒,来确定FTO的去甲基酶活性是否必须。LC-MS / MS确认FTO-WT或FTO-MUT的异位表达对细胞m6A水平的影响。异位表达FTO-WT,但不是FTO-MUT,显着增加LC3-II:I表达,LC3斑点显着增强,自噬体的数量提高。这些结果证明FTO去甲基化的活性是前脂肪细胞中自噬所必需的。
调查FTO通过RNA去甲基化影响ATG5和ATG7表达,异位表达FTO-WT增加ATG5和ATG7的蛋白质和mRNA水平,而ULK1蛋白丰度没有变化。在Atg5和Atg7的3'UTR处发现m6A修饰。发现FTO的敲除增加3T3-L1细胞的m6A水平。基因特异性甲基化RNA免疫沉淀-qPCR(MeRIP-qPCR)表明FTO敲低显着增加Atg5和Atg7 mRNA转录物的m6A水平,但不对Ulk1有影响。评估靶mRNA进行m6A修饰对于FTO介导的基因调控是否必要,3T3-L1细胞中进行双荧光素酶报告和诱变实验。强表达FTO-WT,但不是FTO-MUT,大大促进含有Atg5和Atg7的3'UTR片段的单个报告结构的荧光素酶活性,当m6A位置发生突变时,这种增加被消除,证明FTO通过m6A依赖机制调节ATG5和ATG7的表达。油红O染色分析显示FTO-WT促进脂肪细胞分化和甘油三酯积累,验证去甲基化脂肪形成需要FTO的活性。这些结果说明FTO靶向Atg5和Atg7转录物并以m6A依赖的方式介导他们的表达,进一步调节自噬和脂肪生成。
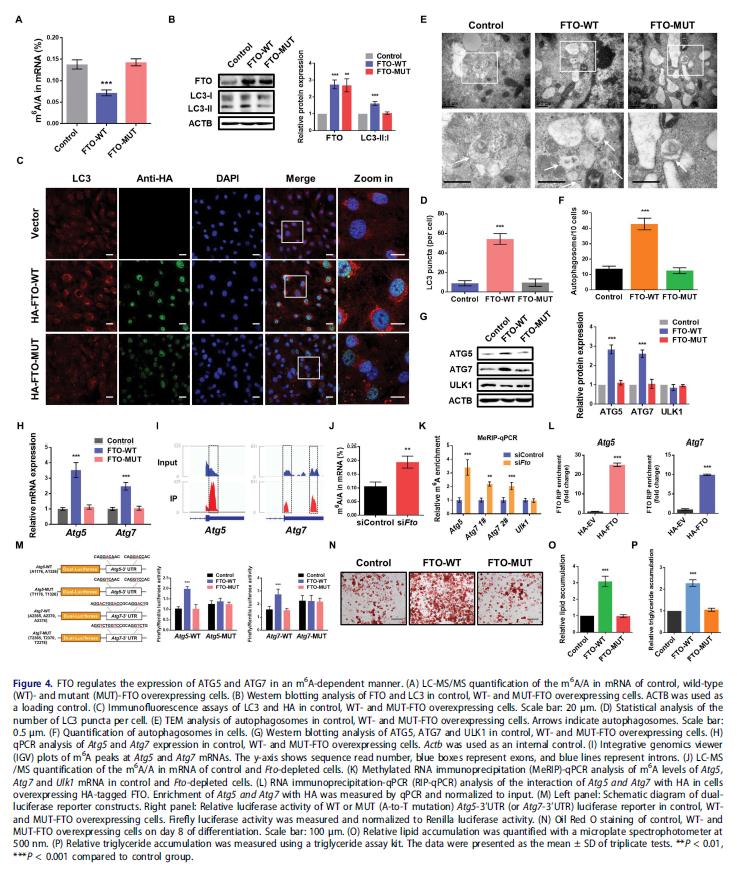
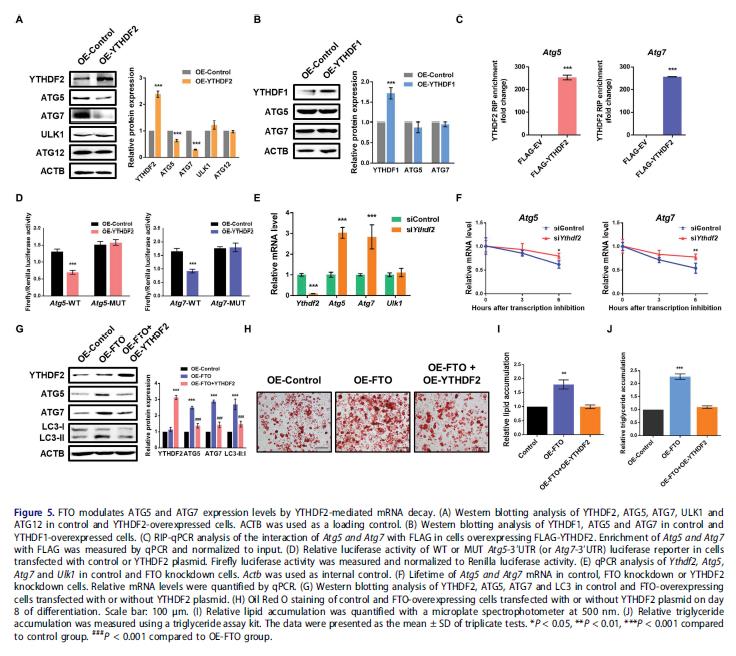
5.YTHDF2通过m6A依赖机制介导ATG5和ATG7的mRNA稳定性和表达
m6A甲基化影响mRNA稳定性和翻译,YTHDF2选择性识别m6A修饰的mRNA并使其不稳定,促进目标mRNA的翻译。进一步解释FTO靶基因的m6A甲基化和蛋白质丰度之间的负相关,首先研究ATG5和ATG7表达是否受YTHDF2或YTHDF1的影响,YTHDF2过表达显着降低ATG5和ATG7蛋白水平,而ULK1和ATG12无变化,强表达YTHDF1不影响ATG5和ATG7的表达。
检测在Atg5和Atg7 mRNAs的m6A修饰对YTHDF2介导的基因调控是是否必需的。YTHDF2异位显着下调野生型Atg5和Atg7片段的荧光素酶活性,这种减少完全被m6A共识位点的突变消除。检测YTHDF2是否介导mRNA衰变从而控制Atg5和Atg7的表达,在3T3-L1细胞中进行功能缺失研究。敲低YTHDF2后 Atg5和Atg7的mRNA水平提高, mRNA稳定性分析显示YTHDF2缺失延长了Atg5和Atg7 mRNA转录物的半衰期。过表达FTO的3T3-L1细胞中ATG5和ATG7蛋白质水平的增加能部分被强表达YTHDF2逆转,LC3-II:I也部分逆转。异位表达YTHDF2可逆转过表达FTO引起的促脂肪生成和甘油三酯积累。这些数据表明FTO调节ATG5和ATG7的表达,通过m6A依赖性和YTHDF2介导的方式。
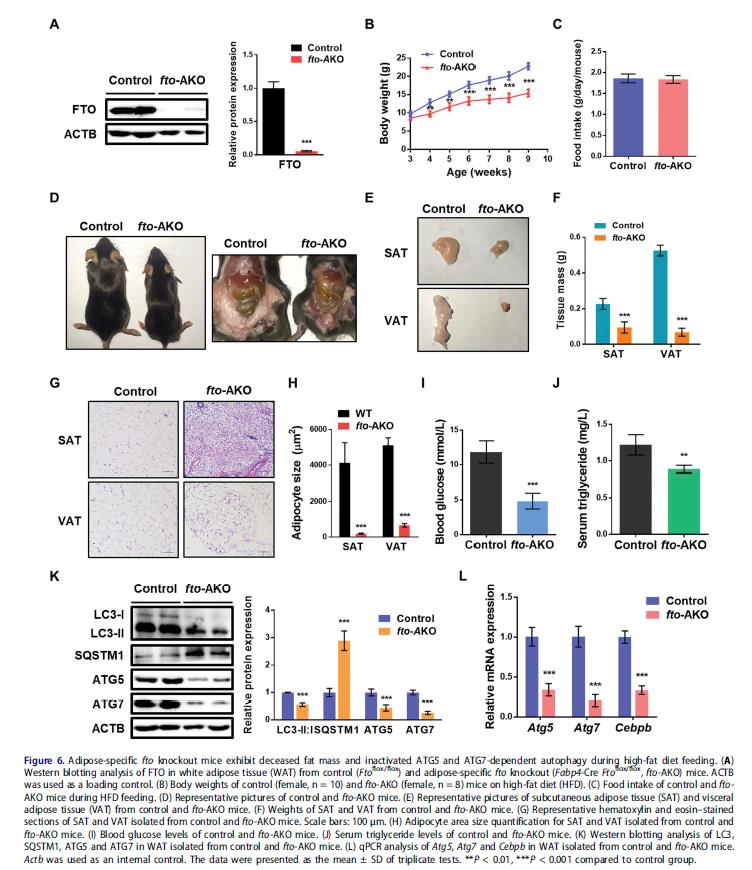
6.脂肪选择性缺失FTO可减少白色脂肪量并抑制ATG5和ATG7依赖性自噬
研究FTO的体内作用,建立脂肪选择性FTO敲除小鼠(fto-AKO)模型,fto-AKO小鼠的脂肪组织(WAT)FTO表达缺失,其他组织包括肌肉,肝脏,大脑,巨噬细胞或血管内皮细胞并没有。相对于对照鼠,食物或高脂饮食(HFD)诱导fto-AKO小鼠的体重增加变弱,且更瘦,而两组之间食物摄入没有显着差异。fto-AKO小鼠腹股沟脂肪和性腺脂肪的数量显着低于同窝对照鼠。SAT和VAT的haematine和伊红染色显示HFD对照小鼠的脂肪细胞表现为一个大脂滴占据整个细胞的典型结构。相反,FTO缺乏导致多腔较小脂滴的脂肪细胞。喂食HFD的fto-AKO小鼠的血糖水平和血清甘油三酯水平的显着低于对照鼠。脂肪选择性缺失Fto减弱LC3-II:I比例和SQSTM1蛋白丰度升高,ATG5和ATG7的基因表达减弱,表明在WAT中自噬抑制。此外, fto-AKO小鼠WAT中Cebpb的mRNA水平下调,暗示fto敲除可能损害脂肪组织发育,通过抑制ATG5和ATG7-CEBPB信号。这些表明脂肪细胞选择性fto敲除可能会减少WAT量和损害ATG5和ATG7依赖性自噬。
总结:
FTO在促进自噬和脂肪生成中起作用,主要通过m6A-YTHDF2依赖机制。这些发现在预防和治疗肥胖的研究进程中迸发出新的火花新的思路。