






载脂蛋白C3在炎症中的新功能
NLRP3-炎症体驱动的炎症与多种疾病的发病机制有关。内源性炎症因子激活剂的鉴定对于开发新的抗炎治疗策略至关重要。德国萨尔兰大学Thimoteus Speer研究组发现,载脂蛋白C3通过替代激活炎性小体诱导炎症和器官损伤。该研究2019年12月9日在线发表在国际学术期刊Nature Immunology:《自然—免疫学》,影响因子= 23.53的一篇论文“Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation”。该研究发现载脂蛋白C3(Apo3)通过胱天蛋白酶-8和Toll样受体2和4的二聚化诱导另一种NLRP3炎症体,从而激活人单核细胞中的NLRP3炎症体。为NLRP3炎症体的调节和含有ApoC3的富含甘油三酯脂蛋白的病理生理学作用提供了新的见解。靶向ApoC3可以防止器官损伤,并为血管和肾脏疾病提供抗炎治疗。

亮点:
(1)该研究发现载脂蛋白C3(ApoC3)通过caspase-8和Toll样受体2和4二聚化的方式诱导NLRP3炎性小体的替代激活,从而激活了人单核细胞中的NLRP3炎性小体。
(2)人单核细胞中炎性小体的替代激活受Toll样受体蛋白SCIMP的调控。这种激活引起Lyn / Syk依赖的钙离子进入和活性氧的产生,导致caspase-8的活化。
(3)在人源化的小鼠模型中,ApoC3在体内以NLRP3和caspase-8依赖性方式激活人单核细胞从而阻止内皮再生并促进肾脏损伤。
结果分析:
一、ApoC3诱导无菌炎症
低密度脂蛋白(LDL)和极低密度脂蛋白(VLDL)处理了人单核细胞,并确定了IL-1β的释放。与HDL和LDL相反,VLDL刺激了IL-1β的释放(图1a)。脱脂的VLDL刺激的IL-1β释放的程度与天然VLDL相似,表明VLDL的蛋白质部分可能是其对IL-1β释放的影响(图1b)。蛋白质组学分析检测到ApoC3,ApoE和ApoC2是VLDL颗粒中三种最丰富的蛋白质。用这些载脂蛋白孵育单核细胞后,发现只有ApoC3才能诱导IL-1β释放(图1c)。六个独立分离的脂蛋白制剂的VLDL级分中检测到大量的ApoC3(图1d)。尽管HDL也包含ApoC3,但它本身并没有诱导IL-1β释放,甚至没有降低ApoC3驱动的IL-1β释放(图1e),这表明HDL及其主要成分ApoA1减少了炎性体的激活,这与之前的研究一致报告。 ApoC3与人单核细胞直接相互作用(图1f)并诱导IL-1β的释放,但也诱导非炎性体依赖性细胞因子白介素6(IL-6)和肿瘤坏死因子(TNF)的浓度依赖性方式(图1g–i)。这表明ApoC3可能激活NLRP3炎性体以诱导IL-1β释放。
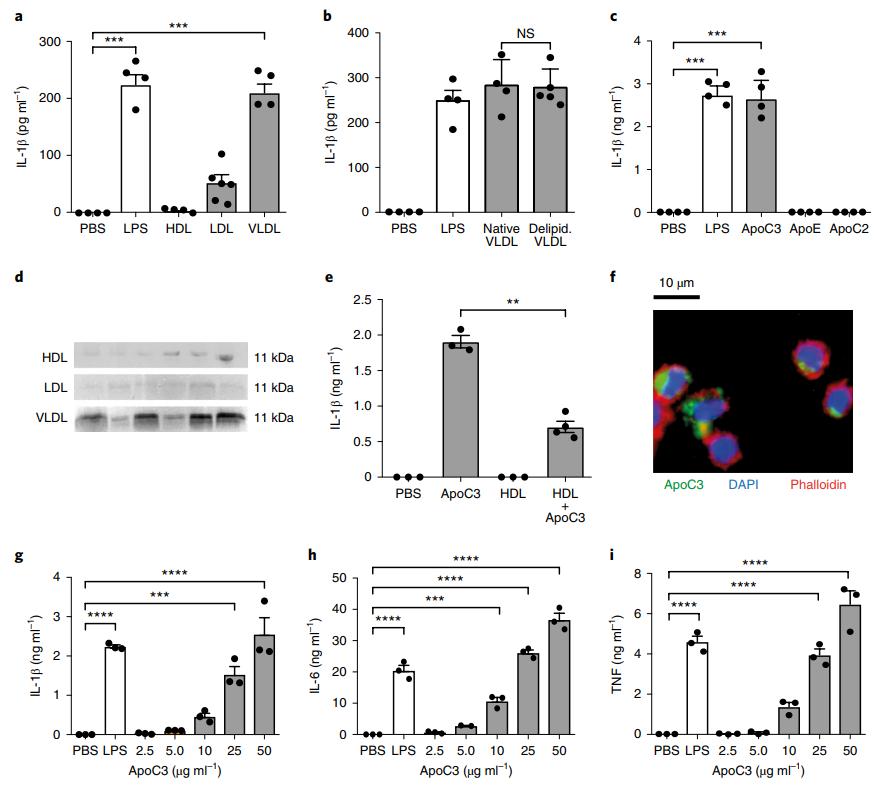
图1:Apo3诱导全身炎症
二、ApoC3激活人类单核细胞中的另一种炎性体途径
ApoC3能够诱导单核细胞释放IL-1β,而无需使用先前报道为生理学相关浓度的TLR配体(图2a)进行预刺激。直接从人血浆中分离的ApoC3诱导的IL-1β释放的程度与整个研究中使用的来自商业供应商的ApoC3的释放程度相似(图2b)。正如预期的那样,ApoC3处理诱导了IL-1β和caspase-1的成熟(图2c)。因此,caspase-1抑制剂Z-YVAD-FMK废除了ApoC3诱导的IL-1β释放(图2d),表明ApoC3释放IL-1β涉及caspase-1的激活。相反,ApoC3仅诱导小鼠骨髓源巨噬细胞(BMDM)中的IL-1β适度释放,与人单核细胞相比降低了十倍(图2e)。ApoC3不会诱导焦磷酸化细胞死亡(图2f)或ASC斑点的形成(图2g)。此外,ApoC3介导的IL-1β释放不依赖于人类单核细胞中的钾流出(图2h)。因此,ApoC3不会诱导加德明D的裂解(图2i)。已经描述了人类单核细胞可以独立于钾外排和热解而响应LPS参与其他的炎性体活化,这需要通过Toll / interleukin-1受体域的适配器诱导干扰素-β(TRIF)受体进行TLR依赖性信号传导-相互作用的丝氨酸/苏氨酸蛋白激酶1(RIPK1)和胱天蛋白酶8。 ApoC3处理诱导了caspase-8的裂解(图2j),并且对caspase-8的催化抑制作用显着降低了ApoC3驱动的IL-1β释放(图2k)。此外,通过特异性抑制剂抑制RIPK1和TRIF可以显着降低响应ApoC3的单核细胞IL-1β释放(图2l)。这些发现表明,ApoC3代表人类单核细胞中替代性炎性体激活的新型诱导剂。
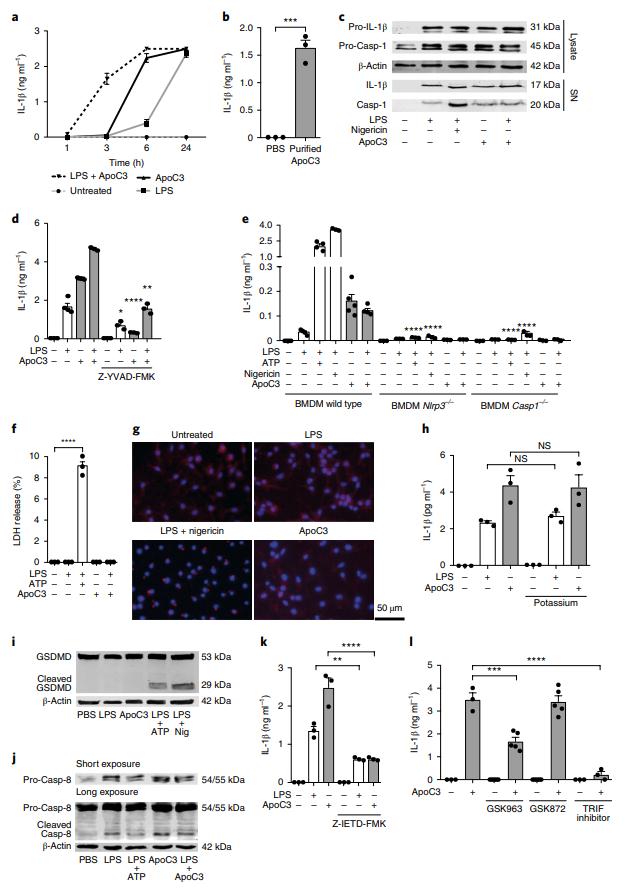
图2:Apo3诱导选择性NLRP3炎症体激活
三、ApoC3需要TLR2和TLR4异二聚体诱导诱导的炎性体激活
NF-κB抑制剂Bay 11-7082完全废除了ApoC3诱导的IL-1β产生(图3a)。细胞外信号调节激酶(ERK)和应激激活的蛋白激酶( SAPKs)/ c-Jun氨基末端激酶(JNK)(图3b)。此外,ApoC3能够引发人类单核细胞进行经典的炎性体激活(图3c)。 ApoC3与被Tlr2或Tlr4相似转染的人类胚胎肾脏(HEK)细胞相互作用(图3d,e)。因此,对TLR2或TLR4的抗体阻断减少了人类单核细胞中ApoC3诱导的IL-1β产生(图3f)。抗体对TLR2或TLR4的阻断降低了ApoC3与人类单核细胞的相互作用(图3g)。为了测试是否需要ApoC3的内在化来诱导IL-1β释放,将ApoC3固定在细胞培养板上,然后将其接种在人单核细胞上(图3h)。接下来,单独或组合用TLR4配体LPS或TLR2配体Pam3CSK4刺激人类单核细胞,以评估每个TLR的个体贡献(图3i)。与单独用LPS处理的细胞相比,用Pam3CSK4刺激LPS引发的细胞可导致更高的IL-1β产生和释放以及caspase-1裂解(图3j)。在未刺激的细胞中,CFP和YFP荧光主要定位在细胞质中(图3k)。这些发现表明,TLR2和TLR4的异二聚体是ApoC3诱导的炎性体激活的关键介质。
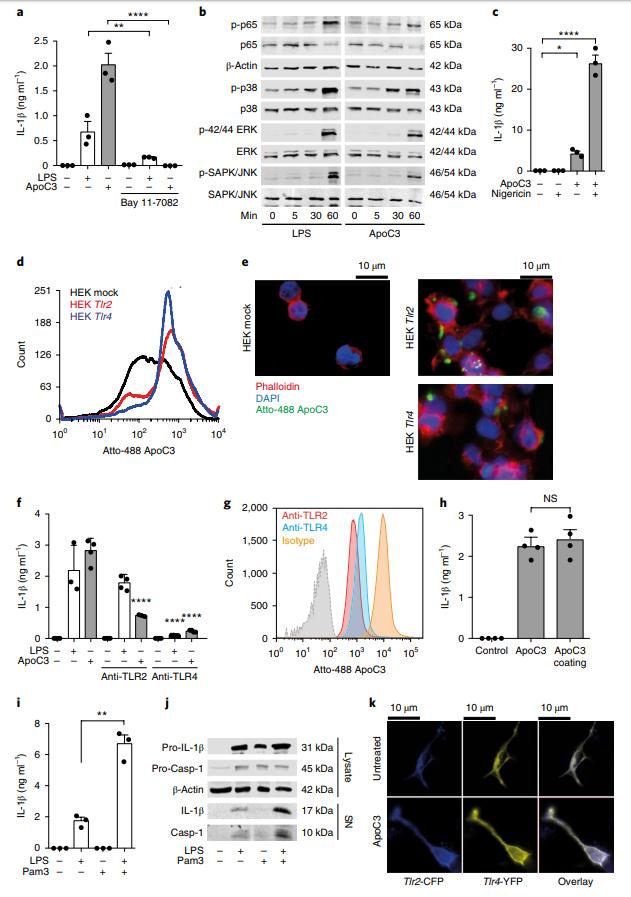
图3:Apo3诱导TLR2和TLR4的异二聚化
四、炎性体的替代活化需要钙依赖性超氧化物的产生
钙离子在ApoC3介导的IL-1β的产生中起着至关重要的作用(图4a)。钙螯合剂还阻止了LPS依赖性IL-1β的产生。因此,ApoC3诱导细胞钙内流(图5c)。瞬时受体电位阳离子通道(TRPM2)抑制剂氟芬那酸,而不是储库操纵的钙进入抑制剂BTP-2或磷脂酶C抑制剂U-73122,显着降低ApoC3诱导的IL-1β释放(图4b)。通过电子自旋共振(ESR)光谱测量,ApoC3和LPS处理均显着增加了超氧化物的产生(图4c,d)。在钙螯合剂存在下,ApoC3诱导的超氧化物产生减少(图4e)。用抗氧化剂N-乙酰半胱氨酸(NAC),黄素蛋白抑制剂DPI或NADPH氧化酶抑制剂VAS-2870预处理单核细胞,几乎完全消除了响应ApoC3和LPS的IL-1β释放,同时阻断了使用MitoTEMPO的线粒体ROS无效(图4f)。该发现表明氧化应激触发了交替的炎性体活化的信号级联反应,从而导致IL-1β的产生。ApoC3增加了单核细胞中TXNIP的表达,这被BAPTA-AM和NAC废除了(图4i)。
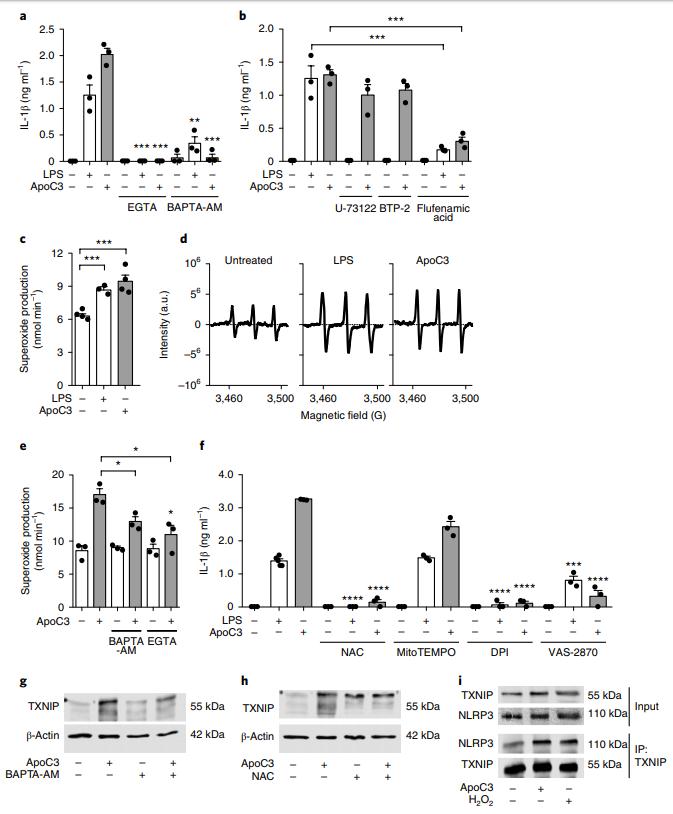
图4:替代性炎症体激活需要钙依赖性超氧化物产生
五、可选的炎性体激活涉及衔接蛋白SCIMP
为了探讨LPS和ApoC3对TLR的激活如何转化为替代的炎症小体激活,集中研究了脾酪氨酸激酶(Syk),它已被证明有助于NLRP3炎症小体激活。ApoC3和LPS触发了Syk的磷酸化(图5a)。因此,抑制Syk可防止LPS和ApoC3诱导的IL-1β释放(图5b)。为了确定Syk是否代表上游信号诱导钙流入和随后的超氧化物产生,在Syk抑制剂R406存在的情况下测量了细胞钙通量(图5c)。Syk的抑制显着降低了人类单核细胞中ApoC3介导的钙内流以及超氧化物的产生和TXNIP表达(图5c-e)。用LPS或ApoC3刺激的人单核细胞中进行了交联实验,并观察到另一个高分子量条带,将其切下并进行了蛋白质组学分析(图5f)。值得注意的是,在用LPS或ApoC3刺激后,检测到了跨膜蛋白SCIMP(图5g)。因此,LPS或ApoC3增加了人单核细胞中SCIMP的表达(图5h)。ApoC3迅速诱导了人类单核细胞中SCIMP,Lyn和Syk之间的相互作用(图5i)。ROS清除钙螯合和Syk抑制减少了LPS响应时前caspase-8的加工,表明SCIMP介导的Syk激活可能代表了caspase-8依赖性替代性炎性体激活的上游事件(图5j– l)。
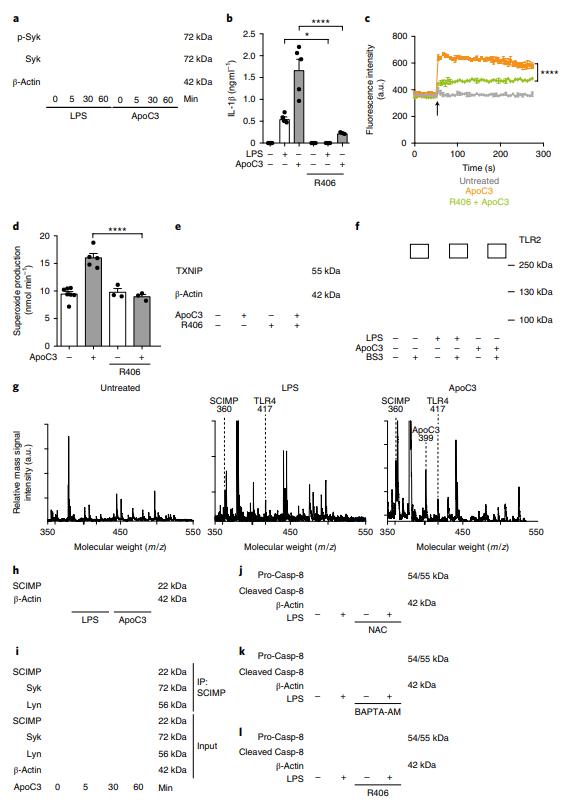
图5:替代性炎症体激活涉及衔接蛋白SCIMP
六、ApoC3在体内诱导人源化小鼠的器官损伤
由于ApoC3激活的其他炎性体激活仅限于人类单核细胞,因此使用了人源化的小鼠模型。通过流式细胞术确定,ApoC3诱导了NOD-SCID小鼠移植人单核细胞中IL-1β的表达(图6a)。 为了评估ApoC3对血管再生的作用,使用血管周围颈动脉损伤模型(图6b)。颈动脉损伤后三天,与用PBS处理的小鼠相比,在用人单核细胞重组并注射ApoC3的小鼠中,受损颈动脉的重新内皮化显着减少(图6c,d)。在用特异性NLRP3抑制剂MCC950或caspase-8抑制剂Z-IETD-FMK治疗的小鼠中,ApoC3对再内皮化的抑制作用消失了(图6e,f)。将用PKH67标记的人单核细胞重构的NOD-SCID小鼠进行单侧输尿管结扎,作为肾脏损伤的模型(图6g)。重要的是,ApoC3强烈增强了肾脏的损害,如扩张的肾小管百分比增加所示(图6h-i)。因此,在经ApoC3处理的小鼠中,在受伤的肾脏中积聚的人类单核细胞数量明显更高(图6j,k)。这些发现表明,ApoC3介导的人类单核细胞活化导致体内器官损伤增加。
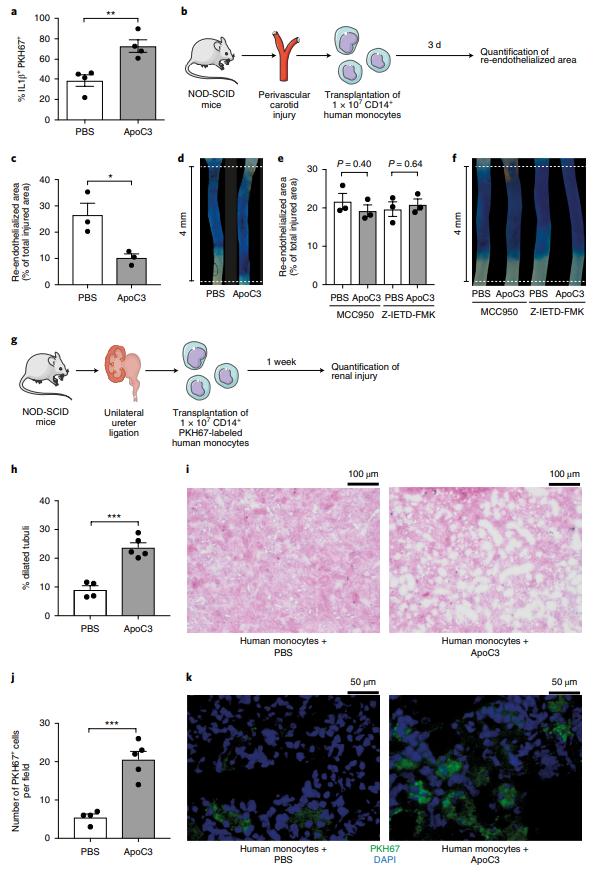
图6:Apo3和人单核细胞在体内诱导器官损伤
七、ApoC3与死亡率增加有关
在急性心肌梗塞(AMI)和CKD患者中测量ApoC3血浆浓度,以强调实验结果的相关性。与健康受试者相比,在患有AMI和CKD的患者中,ApoC3血浆浓度显着更高(图7a,b)。路德维希港风险与心血管健康(LURIC)研究的3,313名参与者进行了冠状动脉造影,其中ApoC3血浆水平高的人具有明显更高的全身炎症标志物,例如E-选择蛋白和高敏感性C反应蛋白(hsCRP)(图7c,d)。为了研究中位9.9年随访期间ApoC3与全因死亡率之间的关联,建立了危险比图(图7e)。值得注意的是,ApoC3血浆水平与更高的全因死亡率显着相关。这突显了ApoC3作为炎症小体驱动的炎症介质的临床意义。
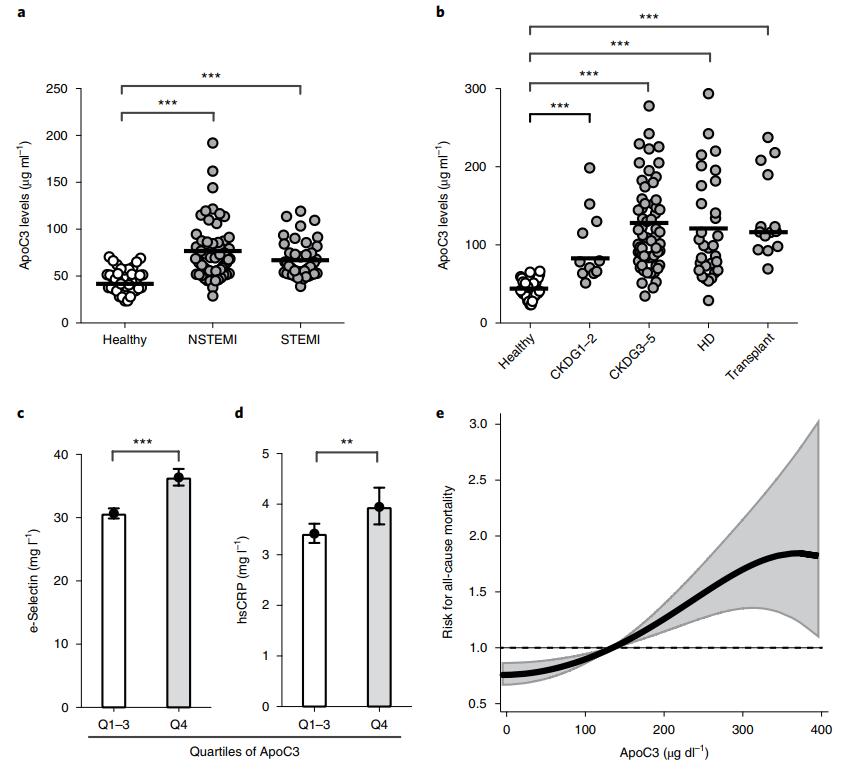
图7:Apo3与死亡率增加相关
结论:
ApoC3是一种新型的介体,通过TLR2,TLR4和SCIMP的异源三聚化以及随后的器官损伤,导致人类单核细胞中的炎性体替代激活。 这些发现不仅与ApoC3有关,而且扩展了对替代性炎症小体激活所需的信号通路的认识。 该研究为NLRP3炎性小体的调控以及富含ApoC3的甘油三酯脂蛋白的生理病理作用提供了新的见解。靶向ApoC3可以防止器官损伤并为血管和肾脏疾病提供了新的抗炎治疗策略。