






靶向疗法导致癌细胞耐药性产生的根本原因!
摘要:
耐药性的出现限制了靶向疗法在人类肿瘤中的功效。普遍的看法是耐药是既成事实:开始治疗时,癌症已经含有耐药性突变细胞。暴露于抗生素的细菌会暂时增加其突变率(自适应变异性),从而提高了生存的可能性。作者调查了人类结直肠癌(CRC)细胞是否同样利用自适应变异性来规避治疗压力。作者发现表皮生长因子受体(EGFR)/ BRAF抑制作用下调错配修复(MMR)和同源重组(HR)DNA修复基因,并同时上调药物耐受性(persister)细胞中易错的聚合酶。在治疗期间,源自患者的异种移植物和肿瘤标本中的MMR蛋白也下调。 EGFR / BRAF抑制可诱导DNA损伤,增加变异性并触发微卫星不稳定性。因此,像单细胞生物一样,肿瘤细胞通过增强变异性来规避治疗压力。

该文验证了一种假说,即在治疗过程中基因组不稳定性的短暂增加也可能促进对靶向疗法的耐药性,从而导致从头诱变。类似的过程,会增加对抗生素产生抗药性的微生物菌株的出现。在稳定的微环境中,微生物的突变率通常很低,从而避免了有害突变的积累。但是,已经在细菌和酵母中描述了几种由压力引起的遗传不稳定性和增加的变异性的机制,称为应激诱变(SIM)。通过降低生长速率,细菌持久性细胞可以在抗生素施加的致命应激条件下生存。
结果一、靶向治疗引起的MMR下调细胞的HR和HR水平
作者研究了微卫星稳定(MSS)人结肠直肠癌(CRC)细胞系对抗EGFR(表皮生长因子受体)抗体西妥昔单抗的反应,西妥昔单抗与panitumumab一起被批准用于治疗肿瘤缺乏RAS和BRAF突变的转移性CRC。接下来,作者评估了药物治疗后CRC细胞是否能调节DNA修复基因的表达。转录谱显示,MMR基因MLH1,MSH2,MSH6以及同源重组(HR)效应子(如BRCA2和RAD51)的表达降低(图1A)。 EXO1(一种编码核酸外切酶的基因,参与失配和双链断裂(DSB)修复)的表达也受到了影响(图1A)。还观察到了MMR和HR蛋白的时间依赖性下调(图1B)。在用靶向药物处理过的CRC细胞中,MMR能力(MMRp)降低至与观察到的水平相当在LIM1215中(图1C)。接下来,作者通过使用基于质粒的两步法pDRGFP / pCBASce-I分析评估细胞的HR能力。在pDRGFP质粒稳定表达后,作者测量了由Sce-1表达诱导的DSB上绿色荧光信号的产生。通过这种测定,用靶向疗法治疗后,DiFi和WiDr细胞均显示出HR能力的显着降低(图1D)。
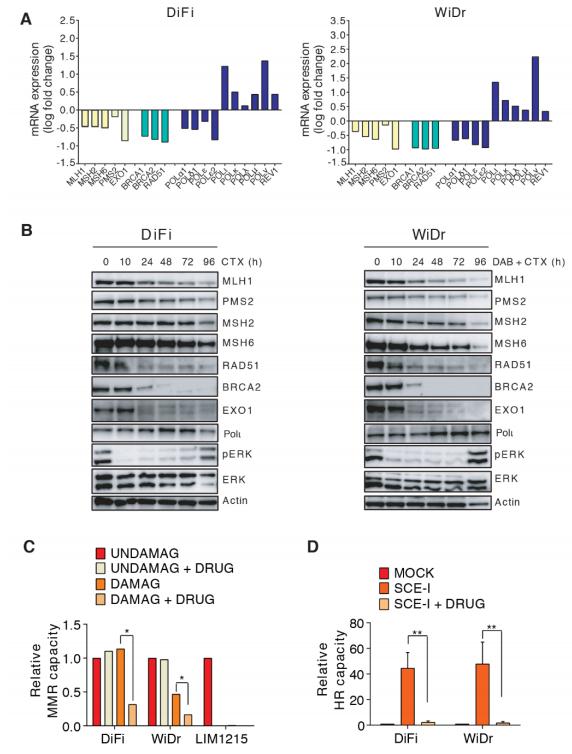
图1. CRC细胞调节DNA修复效应因子以响应靶向药物
结果二、靶向治疗后CRC残留疾病样本中的MMR蛋白下调
为了确定基于细胞的发现是否扩展到患者来源的肿瘤样品,作者选择了六个具有野生型KRAS,NRAS和BRAF的MSS PDX模型,其中西妥昔单抗抑制EGFR导致肿瘤消退的程度不同,与临床情况平行(图2A)。免疫组织化学分析显示,当肿瘤处于对西妥昔单抗的最大反应点时,所获得的所有肿瘤样品中MLH1和/或MSH2的表达均下调,但仍含有残留的持久性物质与安慰剂治疗的对照组相比(图2B和C)。接下来,作者调查了两名CRC患者的临床标本中是否也发生了DNA修复蛋白的下调,这些患者在用FOLFOX加panitumumab治疗后达到了客观的部分应答。在这两种情况下,当尽管治疗仍存在有限数量的肿瘤细胞时,在诊断和最大治疗反应时纵向收集肿瘤标本。与治疗前的标本相比,在应答后获得的肿瘤标本中的MLH1和MSH2下调,证实了作者研究结果的临床意义(图2D)。
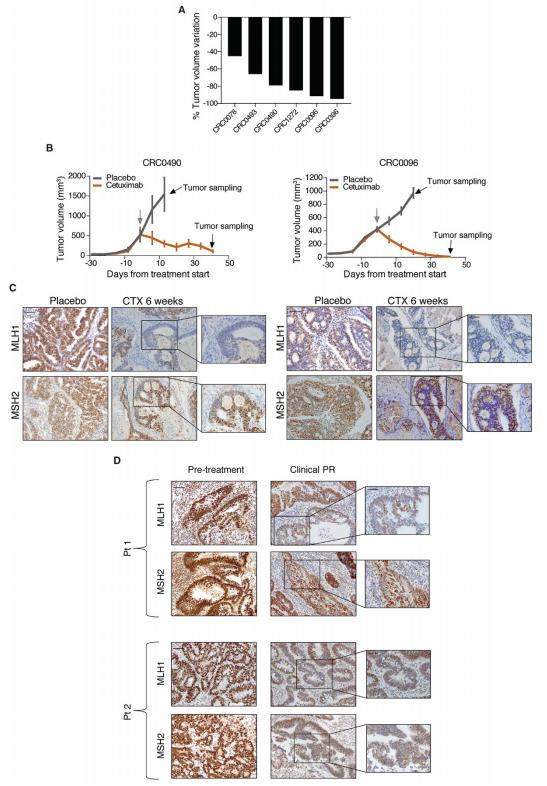
图2. CRC PDXs和靶向治疗患者的MMR下调
结果三、在靶向治疗后的CRC细胞中诱导DNA损伤和易错DNA聚合酶
作者对DNA损伤的常见标记物Ser 139(γH2AX)处H2AX磷酸化的定量分析显示,药物治疗后病灶阳性核的数量呈剂量和时间依赖性增加(图3A-B)。这些数据表明靶向疗法触发了从高保真到易错介导的DNA损伤修复的转变,从而潜在地增加了赋予耐药性的突变的发生。接下来,作者探讨了靶向治疗后观察到的DNA损伤的可能原因。尽管几种化学治疗剂直接产生DNA损伤,但干扰致癌信号的药物(例如EGFR或BRAF抑制剂)并非直接具有遗传毒性。某些靶向疗法会增加癌细胞中的活性氧水平,可能在治疗过程中造成DNA损伤。当CRC细胞暴露于EGFR和BRAF抑制剂时,活性氧水平显着增加(图3C)。当在抗氧化剂N-乙酰基-L-半胱氨酸(NAC)存在下进行靶向治疗时,可以消除药物引起的ROS水平升高(图3C)。
结果四、干扰致癌依赖性可引发CRC的应激反应
在EGFR和BRAF药理阻断后,mTOR效应物pS6K-p70K被下调,其动力学与MMR和HR调节相当(图3D)。但是,mTOR沉默不会影响DNA修复蛋白或γH2AX的表达(图3E)。实际上,DiFi细胞中siRNA介导的EGFR或KRAS的敲低以及WiDr细胞中的BRAF(+/- EGFR)导致DNA修复蛋白表达降低,触发了DNA损伤和mTOR下调(图3E)。这些结果排除了药物诱导的DNA修复途径的下调可能是由于抗EGFR抗体西妥昔单抗或BRAF抑制剂dabrafenib的非特异性(脱靶)作用引起的。
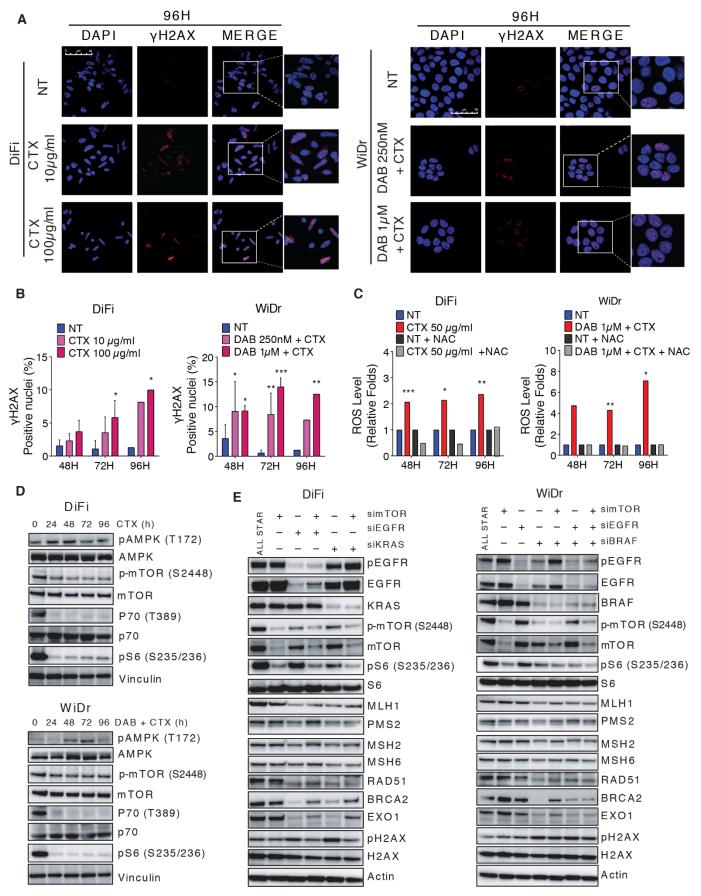
图3. 靶向治疗可触发应激反应,增加ROS水平,诱导CRC细胞DNA损伤
结果五、靶向疗法可诱导CRC细胞适应性变异
二核苷酸CArepeat微卫星驱动了NanoLuc酶编码序列失读(图4A)。为了验证测定,作者首先引入了CA-将NanoLuc载体导入MMRd人类CRC细胞系(HCT116)和三种MMRp人类CRC细胞系(DiFi,WiDr,NCIH508)。在标准生长条件下48小时后,MMRd细胞中的NanoLuc信号明显更高(图4B)。当将HCT116放置在培养物中数天时,这种差异进一步增加,而MMRp品系中的信号仍然很低(图4B),这表明CA NanoLuc分析有效检测了癌细胞中的MMR缺乏症。MLH1 KO克隆表现出预期的更高水平的NanoLuc信号,证实了该测定法可以检测DNA MMR的失活(图4C)。接下来,评估了药物依赖性(瞬态)MMR的下调。 EGFR和BRAF抑制导致生物发光的时间依赖性增加(图4D),与DNA修复效应子的下调和低保真聚合酶的上调平行。
结果六、靶向疗法可诱导CRC细胞适应性变异
作者使用了一种记者分析方法,其中二核苷酸CArepeat微卫星驱动了NanoLuc酶编码序列失读(图4A)。首先引入了CA-将NanoLuc载体导入MMRd人类CRC细胞系(HCT116)和三种MMRp人类CRC细胞系(DiFi,WiDr,NCIH508)。在标准生长条件下48小时后,MMRd细胞中的NanoLuc信号明显更高(图4B)。MLH1 KO克隆表现出预期的更高水平的NanoLuc信号,证实了该测定法可以检测DNA MMR的失活(图4C)。接下来,评估了药物依赖性(瞬态)MMR的下调。EGFR和BRAF抑制导致生物发光的时间依赖性增加(图4D),与DNA修复效应子的下调和低保真聚合酶的上调平行。

图4. 靶向治疗促进CRC细胞的诱变
结果七、靶向治疗后CRC细胞的基因组改变
为了确定在使用EGFR和BRAF抑制剂处理的CRC细胞的基因组中是否存在适应性变异的分子证据,作者分析了DiFi和WiDr亲代,持久性和耐药性衍生细胞的全外显子测序(WES)数据。作者还检测到了对靶标药物具有抗性的CRC细胞微卫星区域的遗传不稳定性增加(图5A和B),突出了靶向治疗对DNA的影响修复过程和致突变性。为了检测非克隆细胞群体中微卫星变化的发生,作者利用了一个高深度捕获板高灵敏度的分析揭示了持久性和耐药性细胞中微卫星区域长度的显着变化(图5C)。对西妥昔单抗耐药的肿瘤组织的WES分析显示,微卫星基因组区域发生了变化,而从相应的未治疗小鼠收集的PDX肿瘤中却不存在这种变化(图5D和E)。总体而言,暴露于靶向疗法的CRC细胞和CRC PDX在核苷酸重复区域中经历了复制保真度的损失。
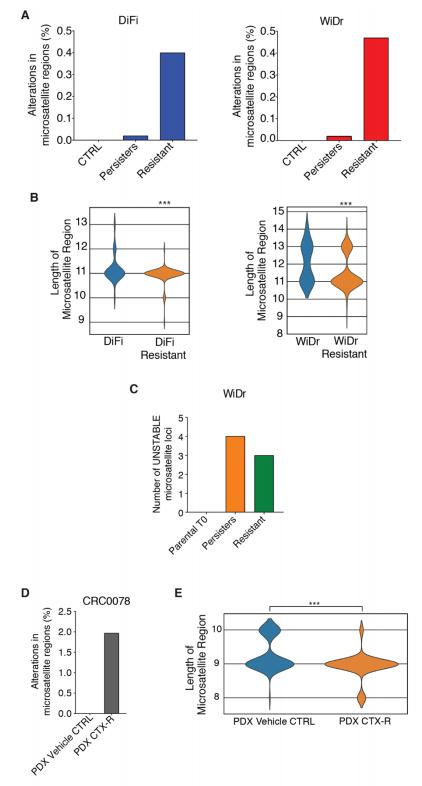
图5. 适应性突变导致CRC细胞在治疗诱导的应激反应中的遗传不稳定性
结论:
EGFR / BRAF抑制可诱导DNA损伤,增加变异性并触发微卫星不稳定性。因此,像单细胞生物一样,肿瘤细胞通过增强变异性来规避治疗压力。可以采用药理学或遗传学上的抑制作用来抑制启动药物驱动的适应性诱变的细胞机制,以减少治疗过程中新变异的产生。这种策略可能会增加和延长靶向疗法的临床疗效。