






m6A甲基化通过FOXO3介导的自噬调节肝癌索拉非尼耐药
m6A是mRNA中一种丰富的核苷酸修饰物,它可以调节mRNA的稳定性、剪接和翻译,但它是否在肿瘤内微环境和肿瘤耐药中也有生理作用尚不清楚。因此小编为大家带来发表于“The EMBO Journal”的文章“RNA m6A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy”,让大家了解m6A甲基化调节肝癌索拉非尼耐药的机制。

结 果:
1.在索拉非尼耐药的肝细胞癌中METTL3下调
为了探讨索拉非尼耐药在肝癌中的分子机制,我们从长期接受索拉非尼治疗的患者中获得了索拉非尼耐药的肝肿瘤,并进行转录组测序。我们发现索拉非尼耐药肝肿瘤中819个基因显著上调,775个基因显著下调。利用KOBAS进行的基因富集分析发现了与耐药性有关的几种信号通路的失调,包括细胞对化学刺激的反应、对有机物的反应和对氮化合物的反应(图1A)。在这四条信号通路中有13个基因重叠(图1B)。我们发现METTL3在索拉非尼耐药肝癌中显著下调(图1C)。通过RT-PCR进一步验证了METTL3在索拉非尼耐药的肝肿瘤中的下调作用(图1D)。通过分析TCGA数据库中肝癌的病理分期图,我们发现METTL3的表达水平在肝癌的晚期显著下调(图1E)。这些结果表明METTL3的下调可能与HCC中的索拉非尼耐药有关。
为了证实METTL3在索拉非尼耐药中的作用,我们系统地分析了GSE62813数据库中METTL3在HepG-2细胞和索拉非尼耐药的HepG-2细胞中表达的变化。我们发现METTL3在索拉非尼耐药肝癌细胞中显著下调(图1F)。通过与原始 HepG-2细胞的比较,我们证实了这些HepG2细胞对索拉非尼的获得性耐药(图1G)。另外,我们发现METTL3在抗索拉非尼的 HepG-2细胞中显著降低,总RNA m6A水平持续下降(图1H和I),表明METTL3在肝癌细胞中介导抗索拉非尼的潜在作用。
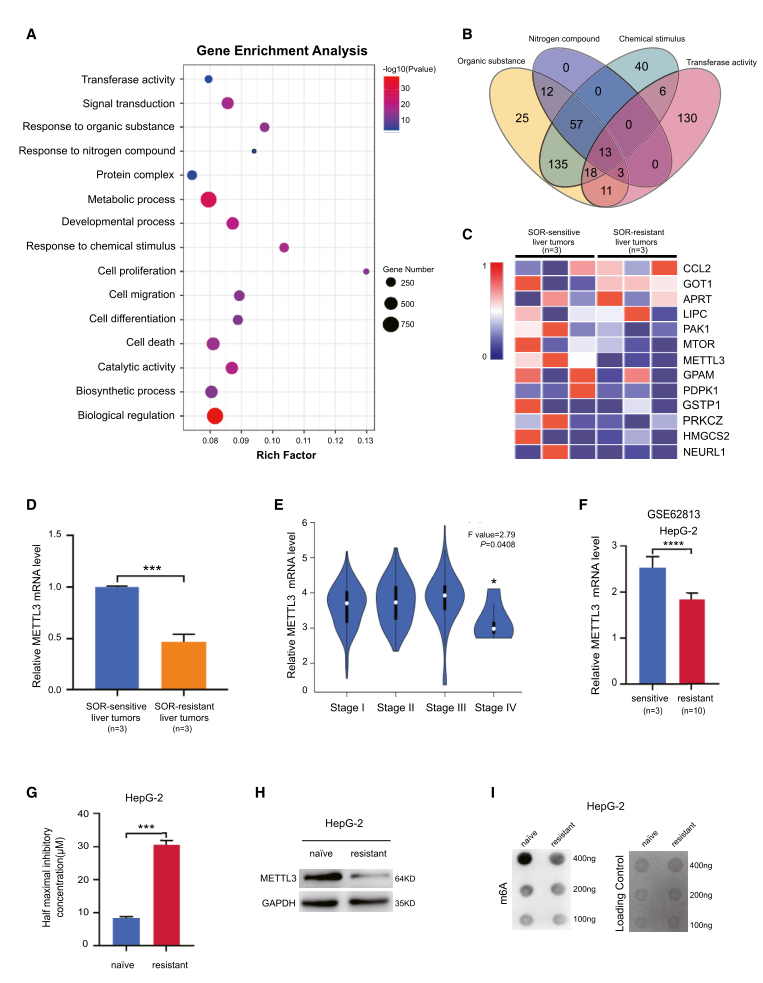
2.METTL3基因敲除增强肝癌中索拉非尼耐药
为了评估整体m6A水平是否与肝癌的发生有关,我们在正常肝细胞系WRL68中产生了一系列不同表达水平的METTL3。正常肝细胞在METTL3沉默后形成一个扩大的集落,但在METTL3过表达后没有明显变化(图2A和B)。METTL3敲除后SMMC-7721、Bel-7402和HepG-2细胞对索拉非尼治疗的敏感性的增加(图2C和D)。此外,为了确定m6A在肝癌索拉非尼耐药中的作用,我们进行了挽救实验(图2E和F)。我们的结果表明,挽救抗shRNA野生型METTL3会使METTL3敲除肝癌细胞和抗索拉非尼HepG-2细胞对索拉非尼治疗敏感(图2G-J),提示METTL3介导的m6A修饰在肝癌索拉非尼耐药中的重要作用。
为了进一步确定METTL3是如何控制肝癌中索拉非尼耐药的,我们测定了METTL3对人脐静脉内皮细胞成管能力的影响。结果显示,与对照组相比,METTL3基因敲除组的肝癌细胞产生的肿瘤条件培养基显示出更强的HUVEC管形成的能力(图2K和K-1)。此外,METTL3基因敲除可上调缺氧条件下肝癌细胞中FGF、PDGF-B、STAT3和VEGF-A等血管生成标志物的RNA表达(图2L和M)。Western blot进一步证实了VEGF-A和PDGF-B的诱导蛋白水平(图2N和O)。综上所述,我们的结果表明METTL3缺失增强了肝癌中的索拉非尼耐药性。

3.METTL3依赖的索拉非尼在肝癌中的耐药是通过促进肝癌的自噬介导的
先前的报告显示,自噬与人类癌症的化疗抵抗有关。为探讨m6A调节是否参与自噬介导肝癌细胞对索拉非尼耐药,采用透射电镜观察了缺氧条件下含或不含METTL3耗竭的SMMC-7721细胞的形态学变化。我们的结果表明,METTL3基因敲除增加了自噬体的数量(图3A)。为了进一步证实这一现象,我们测量了自噬中的生物标志物LC3的脂质水平。结果表明,METTL3基因敲除显著增加了肝癌细胞系中LC3-II的积累(图3B-D)。抗索拉非尼的HepG-2细胞中LC3-II的积累也增强,与亲本细胞相比,其METTL3的表达相对较低(图3E)。值得注意的是,我们通过荧光免疫染色检测到GFP-LC3在METTL3基因敲除的肝癌细胞和索拉非尼耐药的HepG-2细胞中的亚细胞重新分布增加。挽救野生型METTL3减少了GFP-LC3在这些细胞中的积累(图3F-H)。为了探讨自噬在METTL3缺失引起的索拉非尼耐药中的作用,我们测定了IC50。3-MA治疗降低了METTL3基因敲除介导的LC3-II积累(图3I-K),并使3-MA治疗组索拉非尼的IC50显著降低(图3L-N)。这些结果表明METTL3的缺失增加了索拉非尼的自噬通量。
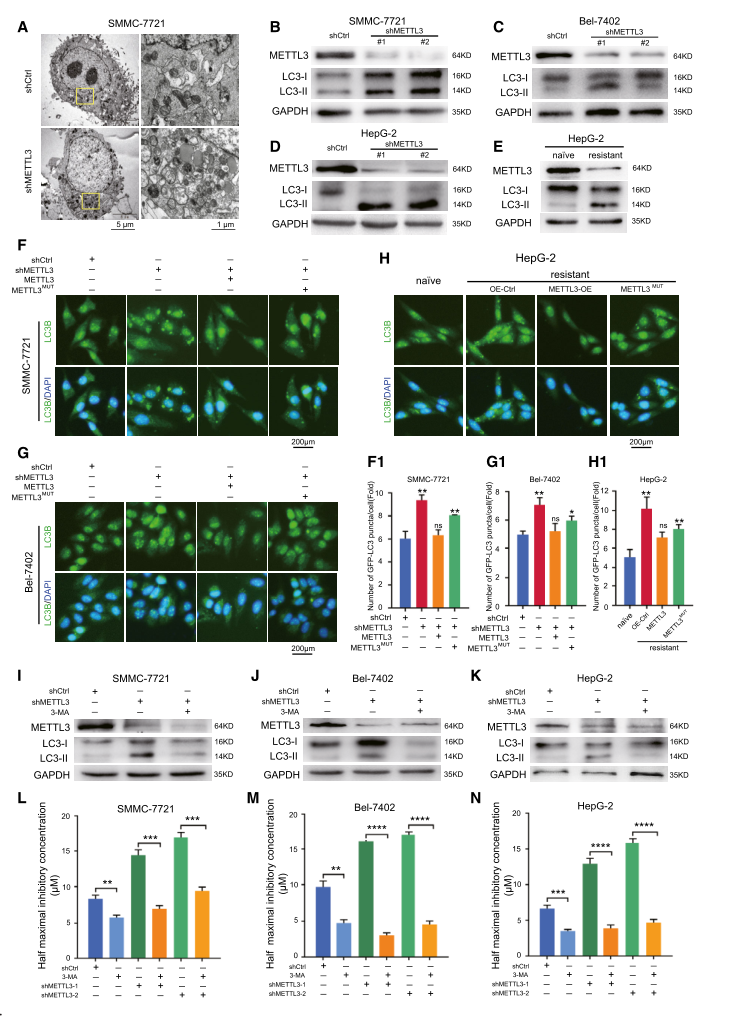
4.METTL3通过介导FOXO3信号调节肝癌细胞自噬
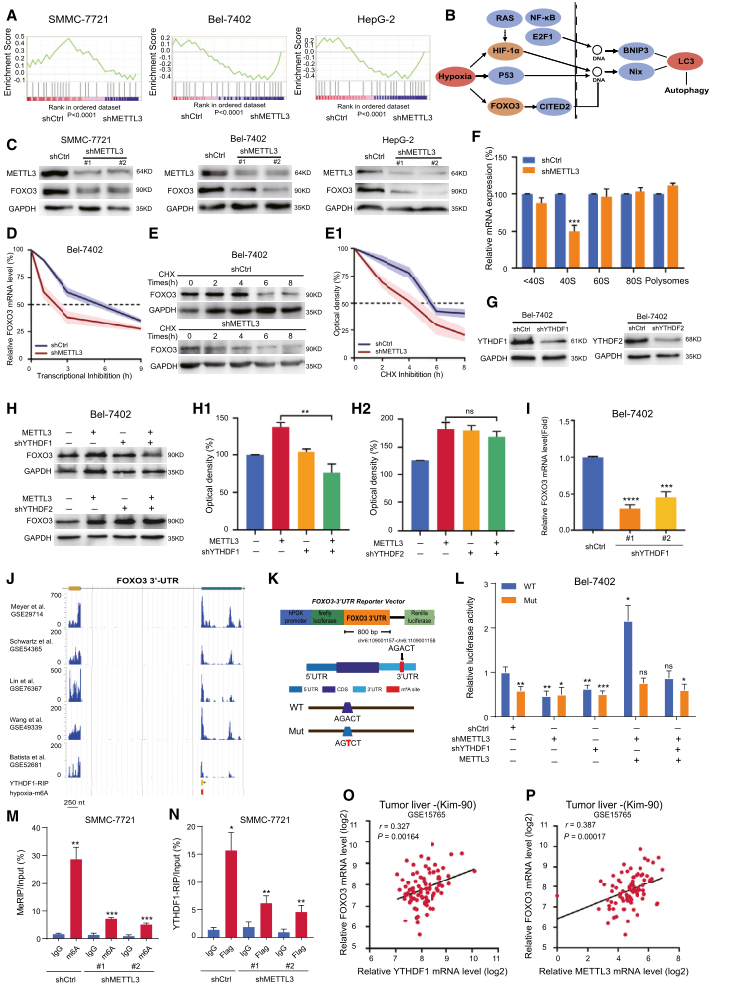
我们探讨了METTL3缺失激活肝癌自噬的机制。基因集富集分析揭示了细胞对逆境基因反应途径的富集。GSEA中富集了23个有显著变化的基因(图4A)。KEGG分析表明,FOXO3是一种普遍的转录因子,当METTL3敲低时,其自噬信号途径发生了显著变化(图4B)。为了确定METTL3如何调控肝癌中的FOXO3,我们首先检测了METTL3基因敲除后FOXO3的表达水平。我们证实在稳定的METTL3敲除细胞中,FOXO3在蛋白质和RNA水平上显著下调(图4C)。此外,经ActinomycinD和Cycloheximide处理后,METTL3基因敲除细胞中FOXO3 mRNA的半衰期和蛋白水平较对照细胞显著降低(图4D和E)。通过RNA组分离,我们发现METTL3敲除显著降低了FOXO3在40S组分下的翻译效率(图4F)。接下来,为了确定调节FOXO3表达水平的潜在m6A读取器,我们在Bel-7402细胞中敲除了YTHDF1和YTHDF2(图4G)。敲除YTHDF1消除了METTL3过表达Bel-7402细胞中FOXO3蛋白水平的升高(图4H)。反过来,YTHDF1的缺失显著降低了FOXO3的RNA表达水平(图4I),表明YTHDF1可能通过m6A修饰参与了FOXO3表达的调控。
为了证明m6A调控对YTHDF1介导的FOXO3表达的影响,我们将FOXO3 3’UTR克隆到一个双荧光素酶报告子结构中,产生FOXO3 3’UTR的突变形式(图4K)。在不同浓度METTL3的Bel-7402细胞中测定了野生型和突变型FOXO3 3’UTR的相对标准化荧光素酶活性。在METTL3敲除和YTHDF1敲除细胞中检测到野生型FOXO3的荧光素酶活性降低。相反,对于FOXO3的3’UTR突变形式,METTL3或YTHDF1的缺失没有任何影响(图4L)。在METTL3过表达的细胞中,YTHDF1的缺失消除了野生型FOXO3对荧光素酶活性的诱导(图4L)。此外,为了验证FOXO3作为缺氧条件下METTL3介导的m6A修饰的真实靶点,我们进行了m6A RNA免疫沉淀分析(MeRIP)和YTHDF1-RIP分析,并通过RT-PCR进行了分析。METTL3的敲除显著降低了FOXO3 mRNA的m6A水平(图4M),并降低了YTHDF1与FOXO3 mRNA的结合(图4N)。此外,通过对肝肿瘤数据集的分析,我们发现在mRNA水平上,YTHDF1与FOXO3(图4O)和METTL3与FOXO3(图4P)呈显著正相关。总之,我们证实FOXO3是METTL3介导的肝癌m6A修饰的关键靶点。
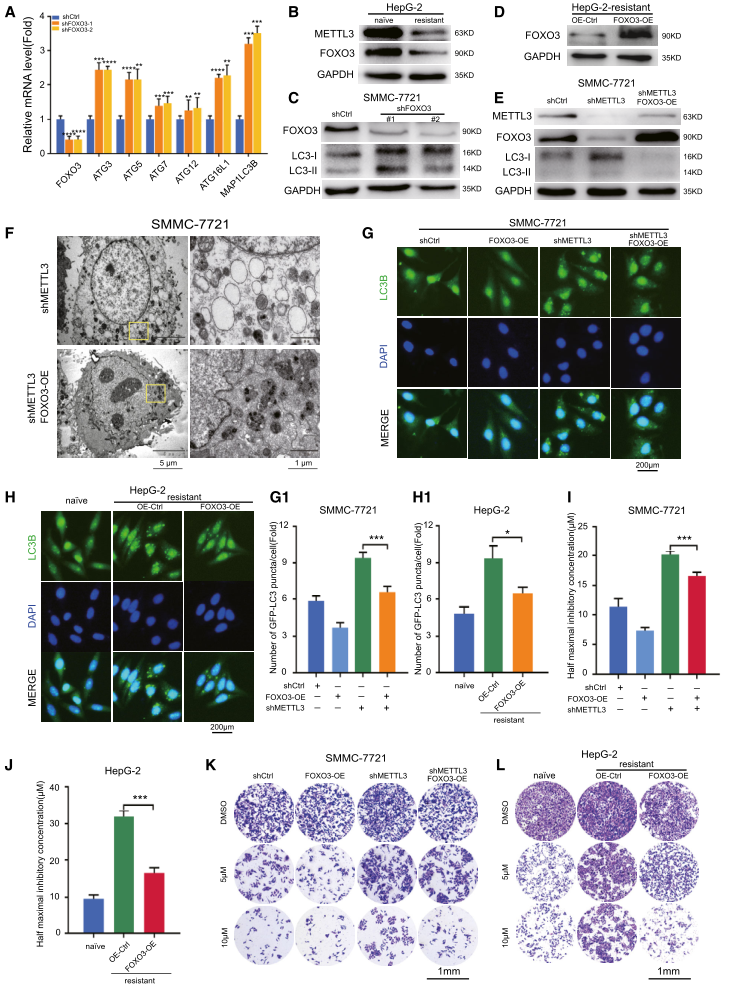
5.过表达FOXO3通过抑制自噬挽救肝癌m6A依赖性索拉非尼敏感性
为了研究FOXO3在自噬信号途径中直接靶点之间的连接性,我们分析了两个数据库,其中FOXO3由强力霉素诱导。FOXO3在自噬中的过表达下调了基因表达谱,而在SMMC-7721细胞中,FOXO3的敲除诱导了自噬相关基因的转录(图5A)。与METTL3的下调一致,FOXO3在索拉非尼耐药的HepG-2细胞中显著下调(图5B)。敲除FOXO3可提高肝癌细胞系的自噬活性(图5C)。为了确定FOXO3是否在METTL3依赖性自噬中起关键作用,我们过表达FOXO3。结果表明,FOXO3在METTL3基因敲除细胞中的过表达降低了自噬活性(图5E),减少了自噬体的数量(图5F),并延缓了GFP-LC3的亚细胞再分布(图5G)。同样,在索拉非尼耐药的HepG-2细胞中过表达FOXO3的荧光免疫染色显示相同结果(图5H)。我们还发现,FOXO3的过表达显著影响了m6A介导的耐药肝癌细胞对索拉非尼的治疗(图5I-L)。综上所述,这些结果表明FOXO3在肝癌中METTL3耗竭介导的索拉非尼耐药中起着关键作用。
6.体内METTL3缺失通过METTL3/FOXO3轴显著增强索拉非尼抗性
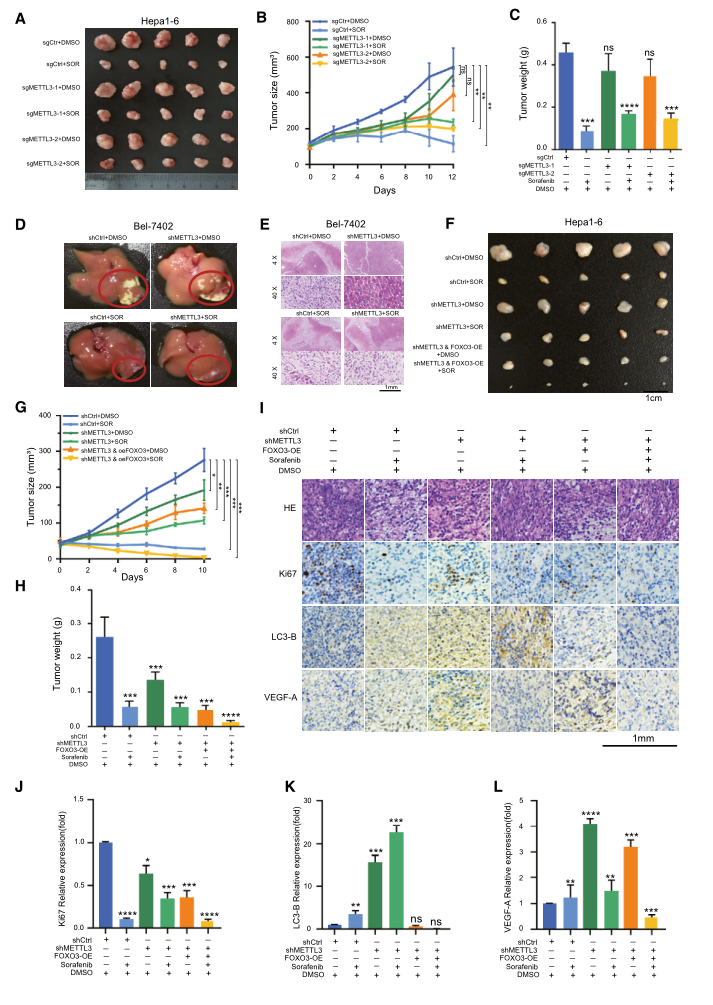
为了验证METTL3在肝癌进展中介导索拉非尼耐药中的作用,我们建立了同种异体移植小鼠模型,并给药:DMSO和索拉非尼(50 mg/kg/2天,腹腔注射)。与我们之前的体外实验结果一致,随着肿瘤大小和肿瘤重量的增加,METTL3基因敲除显著地消除了索拉非尼体内治疗的抑制作用(图6A-C)。此外,我们将Bel-7402细胞直接注射到NOD-SCID小鼠的肝实质中,建立了原位移植性肝癌模型。与对照组相比,METTL3基因敲除消除了索拉非尼对肝肿瘤微环境的抑瘤作用(图6D和E)。为了进一步证实METTL3/FOXO3轴在体内肝癌中的作用,我们构建了同种异体移植小鼠模型,并像以前一样开始给药。一致地,METTL3的敲除显著地消除了索拉非尼在体内的抑制作用。值得注意的是,在METTL3敲除的Hepa1-6细胞中过表达FOXO3挽救了索拉非尼敏感性效应,这反映在与溶剂治疗组相比,肿瘤大小和肿瘤重量显著减少(图6F-H)。与单独使用METTL3相比,在索拉非尼治疗的METTL3敲除的细胞中过表达FOXO3显示出增殖、自噬和血管生成减少(图6I-L)。
此外,我们在BALB/C裸鼠中建立了两个肝癌患者来源的异种移植(PDX)模型,并用四种方案给药(图7A)。与索拉非尼治疗对照组(图7B-D和I)相比,METTL3损耗显著地挽救了异种移植瘤的生长,后者分别表现为Ki67(图7F)、LC3-B(图7G)和VEGF-A(图7H)所示的增殖、自噬和血管生成增加(图7E)。综上所述,我们得出结论:体内METTL3缺失主要通过METTL3/FOXO3轴显著增强索拉非尼抗性。

结 论:
我们的工作揭示了METTL3介导的m6A修饰在缺氧肿瘤微环境中的关键作用,并确定FOXO3是m6A修饰在肝癌抗索拉非尼治疗中的重要靶点。