






PGC1α抑制肝癌转移
PPARγ协同激活因子-1α(PGC1α)是线粒体生物发生和呼吸的关键调控因子。PGC1α参与肿瘤的发生、发展和代谢状态。然而,其在肝细胞癌(HCC)进展中的作用尚不清楚。今天小编为大家介绍发表于“Hepatology”的文章“PGC1α suppresses metastasis of HCC by inhibiting Warburg effect via PPARγ-dependent WNT/β-catenin/PDK1 axis”,带大家了解其中的机制。

在本研究中,我们观察到PGC1α在人肝癌中下调。临床研究表明,PGC1α的低表达与生存率低、血管侵犯和肿瘤体积大有关。PGC1α抑制肝癌细胞在体外和体内的迁移和侵袭。PGC1α通过下调WNT/β-catenin途径介导的PDK1而抑制Warburg效应,PPARγ激活则抑制WNT/β-catenin途径。
结 果:
1.PGC1α低表达与肝癌预后不良相关
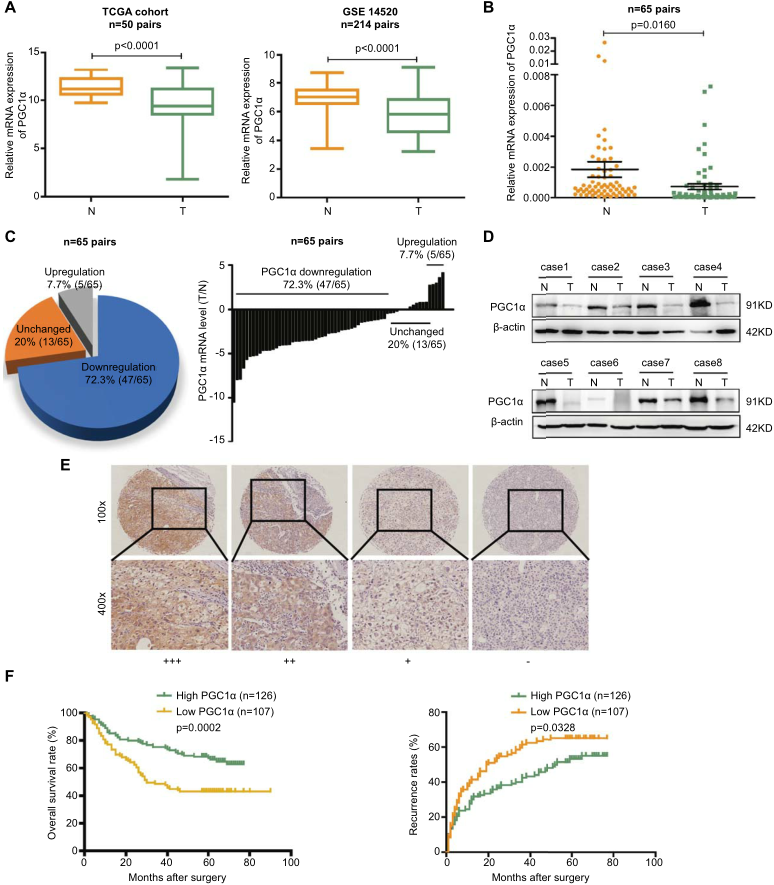
通过TCGA和GSE14520队列分析,发现PGC1α在肝癌中下调(图1A)。采用实时PCR技术检测肝癌组织及癌旁非癌组织中PGC1α的表达水平。如图1B-C所示,与相应的非肿瘤肝组织相比,肿瘤组织中PGC1α的mRNA水平降低了72.3%。western blot进一步证实了PGC1α的下调(图1D)。接着,为探讨PGC1α在肝癌组织中下调的临床意义,对肝癌患者的组织芯片进行了免疫组化染色。PGC1α的代表性免疫染色如图1E所示,根据免疫组化结果将肝癌患者分为两组:PGC1α高表达组和PGC1α低表达组。Kaplan-Meier生存分析显示,PGC1α表达水平低的肝癌患者总生存率(OS)和复发时间低于PGC1α水平高的患者(图1F)。总之,这些数据表明PGC1α可能是预测肝癌患者预后的一个有价值的因素。
2.PGC1α抑制肝癌细胞体内外转移
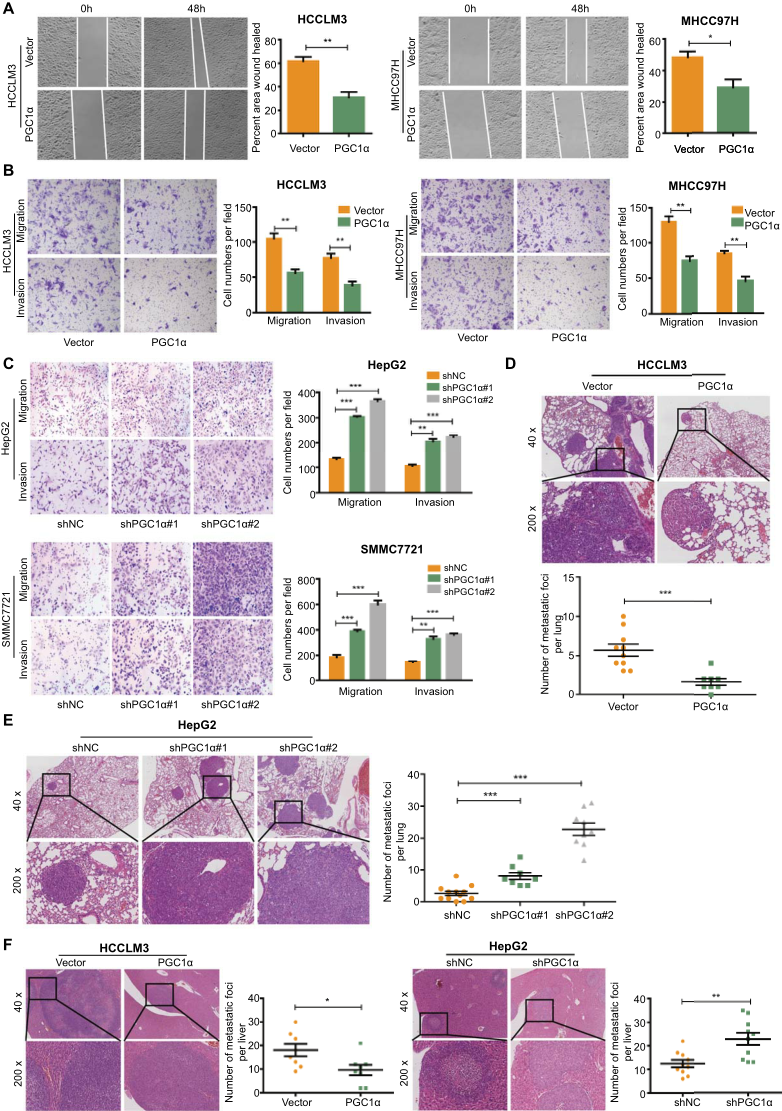
为了探讨PGC1α在肝癌细胞中的生物学功能,我们建立了PGC1α在HCCLM3和MHCC97H细胞系中高表达的稳定模型,并根据PGC1α在肝癌细胞中的表达水平,建立了PGC1α在HepG2和SMMC7721细胞系中低表达的稳定模型。迁移和侵袭试验表明,PGC1α的过表达显著抑制了HCC细胞的迁移和侵袭性(图2A-B),而PGC1α的敲除显著促进了HCC细胞的迁移和侵袭性(图2C)。此外,为了在体内证实上述发现,我们将稳定的细胞株注入裸鼠的侧尾静脉,建立肺转移模型。8周后,与对照组相比,注射PGC1α高表达HCCLM3细胞的小鼠肺转移结节较少(图2D)。相反,PGC1α的敲除显著增加了肺转移结节的数量(图2E)。此外,与对照组相比,PGC1α的稳定过表达显著降低了原位肝癌种植模型中肝内转移灶的数量(图2F)。相反,PGC1α敲除增加了肝内和肺转移结节的数量。这些结果提示PGC1α在体内外均能抑制肝癌细胞的迁移和侵袭,可能在肝癌的发展和转移中起到抑癌作用。
3.PGC1α通过调节Warburg效应抑制肝癌的进展
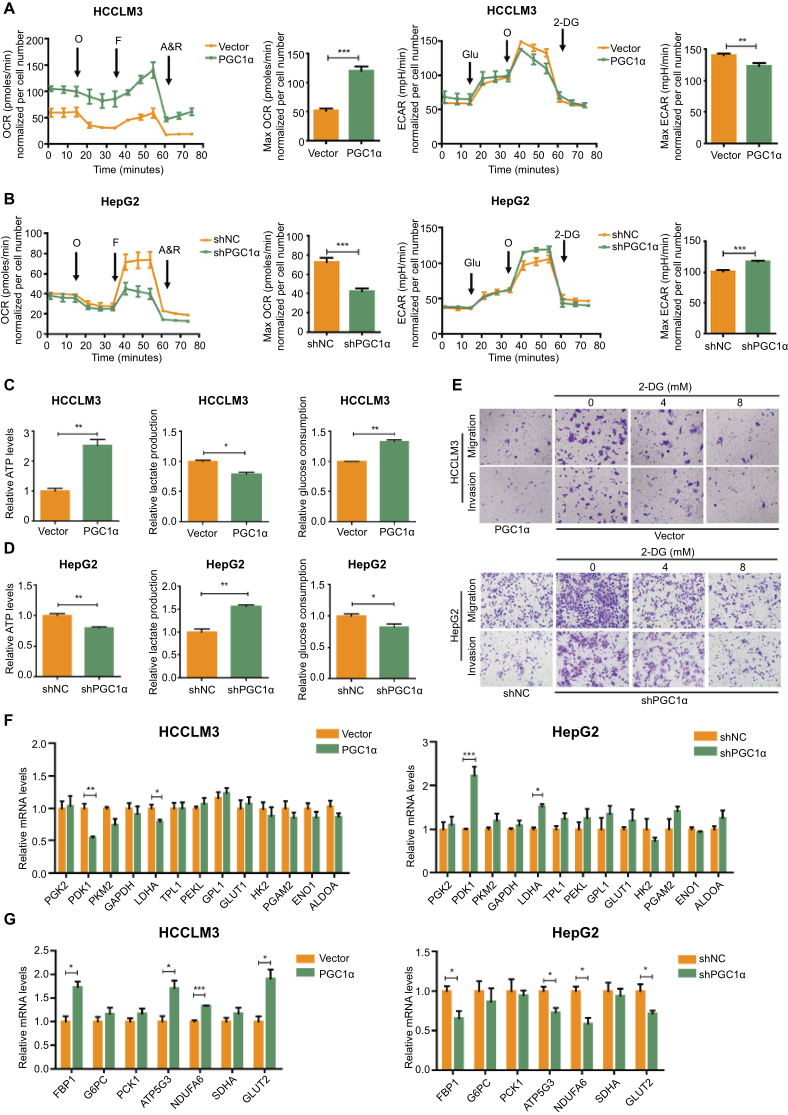
据报道,PGC1α在几种癌症的发展过程中参与了重编程代谢。我们假设肝癌细胞中PGC1α的抑瘤活性伴随着一个整体的代谢重编程。我们比较了PGC1α高表达或基因敲除的肝癌细胞和对照细胞之间的关键细胞代谢和生物能量参数。结果表明,PGC1α的过表达显著提高了HCCLM3和MHCC97H细胞的基础耗氧率和最大耗氧率(OCR),降低了细胞外酸化率(ECAR)(图3A)。相反,PGC1α的敲除降低HepG2和SMMC7721细胞的OCR和ECAR(图3B)。此外,PGC1α的过表达导致细胞ATP水平、葡萄糖摄取量的增加以及细胞外乳酸水平的降低(图3C),而PGC1α的敲除降低了细胞内ATP水平和葡萄糖摄取量,并增加了细胞外乳酸水平(图3D)接着,为了探讨Warburg效应是否与HCC细胞的进展有关,HCCLM3载体和HepG2-shPGC1α细胞分别用不同浓度的2-DG处理,结果显示2-DG对HCCLM3载体和HepG2-shPGC1α细胞的糖酵解有明显的抑制作用。HCCLM3载体和HepG2-shPGC1α细胞的迁移和侵袭能力也呈剂量依赖性下降(图3E)。最后,为了进一步探讨PGC1α调节有氧糖酵解的机制,我们评估了PGC1α对重要糖酵解酶表达的影响。我们用RT-PCR检测了13种糖酵解酶的mRNA水平,发现除PDK1和乳酸脱氢酶(LDHA)外,大多数酶的表达水平没有改变,PDK1的表达水平也基本改变(图3F)。糖异生、氧化磷酸化和肝脏特异性葡萄糖转运蛋白的标记物也通过RT-PCR进行测量(图3G)。综上所述,这些结果提示PDK1是PGC1α调节有氧糖酵解的功能下游靶点,对PGC1α介导的肿瘤进展至关重要。
4.PGC1α对PDK1的抑制作用是由WNT/β-catenin信号转导介导的
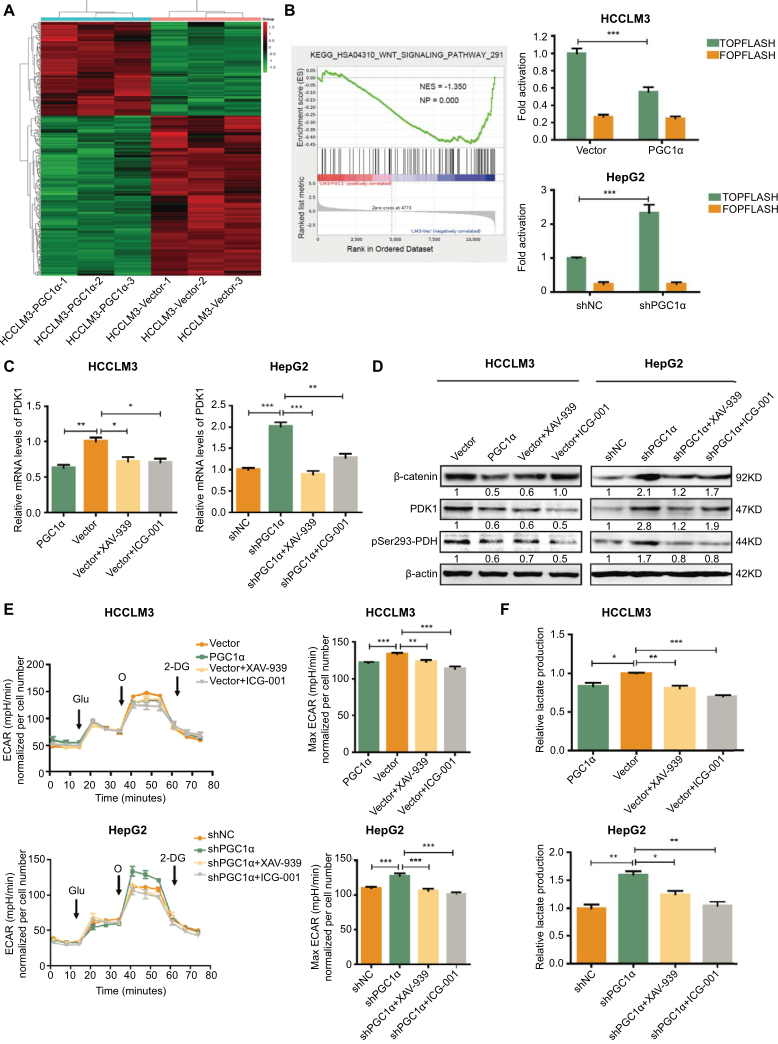
我们研究了PGC1α抑制PDK1的机制。通过RNA-seq评估HCCLM3细胞的整体基因表达模式(图4A)。通过基因集富集分析(GSEA)研究转录组变化对生物功能和途径的影响。我们发现WNT信号通路与HCCLM3细胞中的PGC1α显著负相关(图4B)。采用TOP/FOPFLASH报告分析PGC1α表达对肝癌细胞WNT/β-catenin通路的影响。我们发现PGC1α的过表达显著抑制了HCCLM3细胞中β-catenin的转录活性,而PGC1α敲除后HepG2细胞中TOPFLASH报告活性显著增强(图4B)。WNT/β-catenin信号转导通过PDK1促进结肠癌细胞有氧糖酵解。因此,我们在WNT/β-连环蛋白途径抑制剂XAV-939或ICG-001存在下检测PDK1和β-连环蛋白的水平。如图4C和D所示,XAV-939或ICG-001逆转了HCCLM3载体和HepG2-shPGC1α细胞中PDK1的mRNA和蛋白水平的升高。Pyruvate dehydrogenase(pSer293 PDH)作为PDK1的靶点,其升高水平也被WNT/β-catenin途径抑制剂抑制。β-catenin蛋白水平随着PGC1α的过表达而降低,随着PGC1α的敲除而升高。因此,XAV-939和ICG-001均能抑制HCCLM3载体和HepG2-shPGC1α细胞ECAR和乳酸生成的增强水平(图4E-4F)。这些数据表明PGC1α通过抑制WNT/β-catenin信号通路抑制肝癌细胞PDK1的表达。
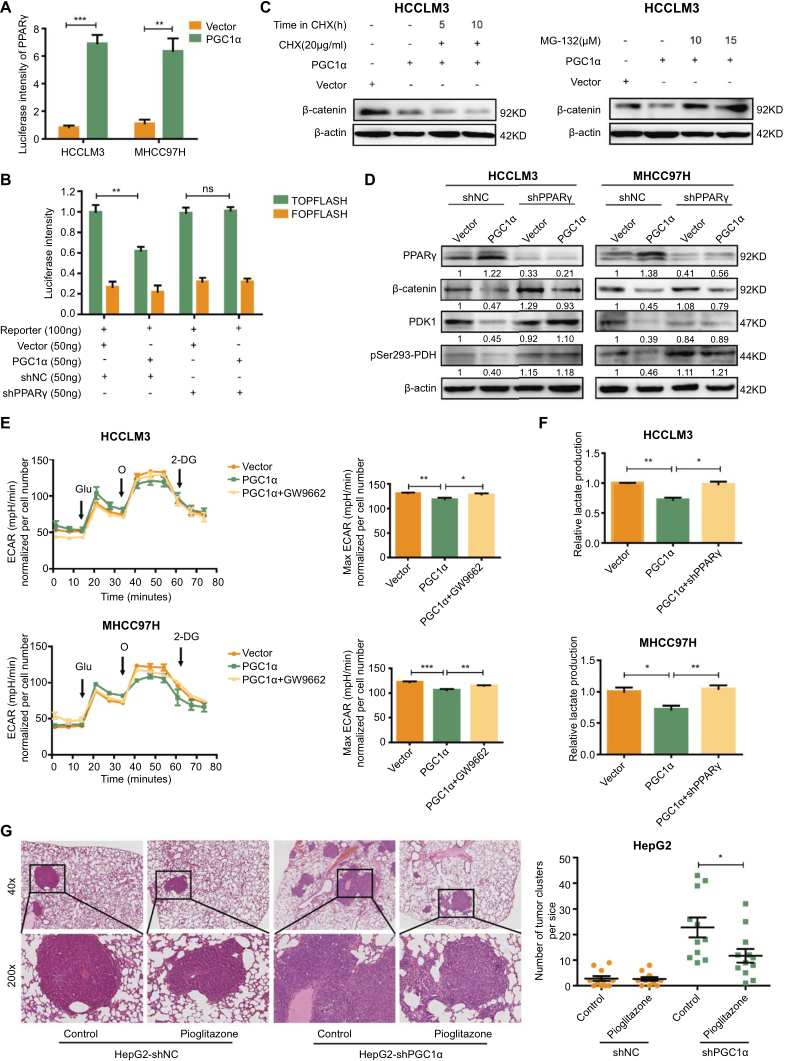
5.PPARγ对PGC1α诱导的WNT/β-catenin通路的抑制和肝癌细胞Warburg效应的影响
PPARγ诱导典型WNT/β-catenin途径的抑制,促进葡萄糖稳态。因此,我们探究PGC1α诱导的WNT/β-catenin信号抑制和抗Warburg效应是否是通过PPARγ依赖方式介导的。双荧光素酶报告分析显示,PGC1α过表达的HCCLM3和MHCC97H细胞中PPARγ的转录活性显著上调(图5A)。采用TOP/FOPFLASH报告分析PPARγ对PGC1α过表达诱导的HEK293T细胞WNT/β-catenin通路的抑制作用。PPARγ基因敲除逆转PGC1α诱导的WNT/β-catenin信号传导抑制(图5B)。这表明PGC1α对肝癌细胞WNT/β-catenin信号传导的抑制作用依赖于PPARγ。
接下来,我们探讨PPARγ如何介导PGC1α诱导的肝癌WNT/β-catenin通路的抑制。结果表明,PGC1α即使在CHX存在的情况下也能降低β-catenin蛋白的水平(图5C,左),表明β-catenin的衰减是一种翻译后机制。然后,用蛋白酶体抑制剂MG-132处理HCCLM3细胞,我们发现PGC1α介导的β-连环蛋白降解被MG-132拮抗(图5C,右),这表明PGC1α介导的β-连环蛋白降解是由HCC细胞中的蛋白酶体介导的途径引起的。此外,通过PPARγ基因敲除,PGC1α诱导的总β-连环蛋白和核β-连环蛋白下调被减弱(图5D)。这表明PGC1α诱导β-catenin降解依赖于PPARγ。
为了探讨PGC1α诱导的抗Warburg作用是否依赖于肝癌细胞中的PPARγ,我们用PPARγ抑制剂GW9662治疗肝癌细胞。如预期的那样,GW9662逆转了肝癌细胞中PGC1α过表达引起的ECAR下降(图5E)。PPARγ的敲除逆转了PGC1α介导的β-catenin、PDK1和pSer293 PDH的下调(图5D),以及细胞外乳酸水平的降低(图5F)。此外,PPARγ激动剂吡格列酮治疗显著抑制PGC1α基因敲除组在体内外的肿瘤进展(图5G)。这些数据表明PPARγ通过增加β-catenin在肝癌细胞中的降解,介导PGC1α诱导的WNT/β-catenin信号传导抑制和肿瘤进展。
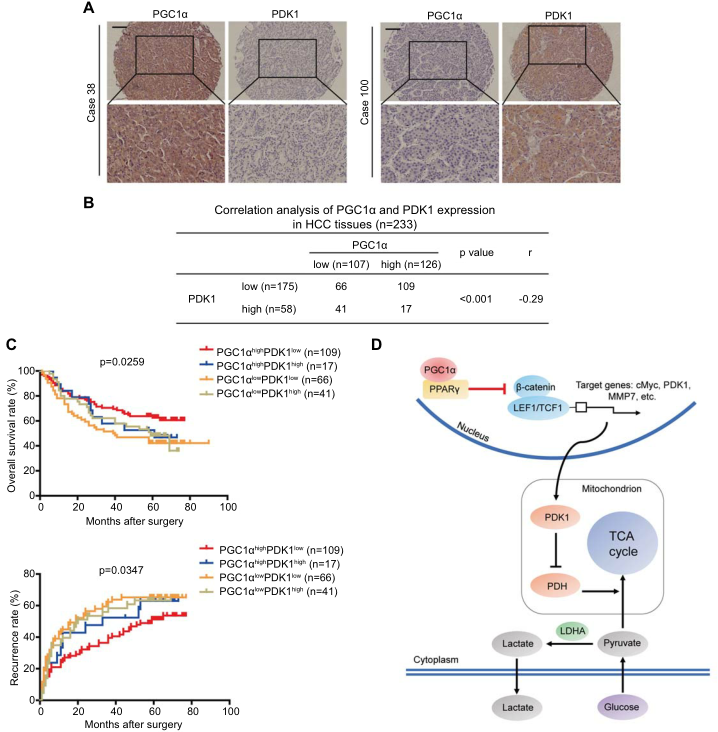
6.肝癌组织中PGC1α与PDK1呈负相关
为了探讨PDK1在HCC中的预后价值,我们分析了肝癌手术切除患者中PGC1α和PDK1的表达。肝细胞癌组织中PGC1α和PDK1的代表性照片如图6A所示。PGC1α高表达的肝细胞癌组织中PDK1的染色较弱,而PGC1α低表达的肝细胞癌组织中PDK1的染色较强。我们分析了肝癌组织中PGC1α表达与PDK1表达的关系。PGC1α表达与PDK1表达显著负相关(图6B)。我们还分析了肝癌组织中PGC1α和PDK1表达水平的预后价值。Kaplan-Meier分析显示PGC1α高表达和PDK1低表达的肝癌患者OS最高,复发率最低(图6C)。这些结果提示PDK1在PGC1α介导的肝癌进展中起作用。
结 论:
我们研究了PGC1α在肝癌转移和代谢中的作用及其机制。PGC1α通过抑制WNT/β-catenin途径下游靶点PDK1的有氧糖酵解抑制肝癌细胞的体内外转移。PGC1α对WNT/β-catenin通路的抑制作用依赖于PPARγ。我们的发现阐明了PGC1α作用的新见解,并提示PGC1α可能是肝癌新治疗策略的一个有希望的靶点。