






mTOR介导的癌症耐药抑制自噬并产生可给药的代谢脆弱性

近日Philipps-University 的Michael Wanzel和Thorsten Stiewe等人将mTOR诱导的肿瘤耐药与自噬缺陷的代谢障碍联系起来,并为治疗难治性肿瘤患者打开了治疗窗口。相关论文于2020年9月份发表在《nature communications》杂志上。
技术路线:

一、顺铂耐药的肺癌代谢脆弱性

我们使用krasq61h突变、p53野生型非小细胞肺癌(NSCLC)患者的H460细胞,将亲代H460 (H460par)细胞通过剂量递增至NSCLC一线化疗标准成分CDDP,获得耐CDDP的H460res细胞。当检测耐药模式时,H460res细胞不仅对CDDP耐药,而且对许多其他破坏DNA的化疗药物也耐药,包括其他铂化合物、etoposide和doxorubicin(图1a)。
相比之下,代谢抑制剂2DG和DCA通过诱导细胞凋亡,严重抑制了H460res细胞的增殖,尽管这两种化合物对亲代细胞的影响不显著(图1a d)。
2DG和DCA目标葡萄糖代谢:2 dg竞争性抑制己糖激酶减缓葡萄糖吸收,而DCA目标抑制丙酮酸脱氢酶激酶,从而减少乳酸生产从糖酵解和促进线粒体丙酮酸的氧化代谢。对来自EGFRL858R、T790M和TP53R273H突变型NSCLC的H1975细胞进行实验,以提高CDDP的剂量(图1e)。
与亲代H1975细胞相比,使用2DG或DCA处理时,CDDP耐药H1975细胞的克隆生长明显减弱,凋亡增加(图1f, g)。
为了验证这些发现是否适用于CDDP治疗后体内复发的肿瘤细胞,我们使用了有条件的KrasLSL-G12D/+;Trp53flox/flox小鼠。吸入后Cre腺病毒,这些老鼠发展成肺腺癌(KrasG12D / +; Trp53Δ/Δ),最初对CDDP治疗但最终复发(图1h).从复发小鼠中获得原发肿瘤细胞培养物,与CDDP幼稚小鼠的肿瘤细胞相比,对CDDP的抗性显著增强,但对2DG和DCA过敏(图1h, i)。
总之,在具有不同驱动癌基因和p53共突变的多种不同肺腺癌模型中,CDDP耐药的发生明显易受糖代谢紊乱的影响。
二、代谢脆弱性是由mTOR介导的
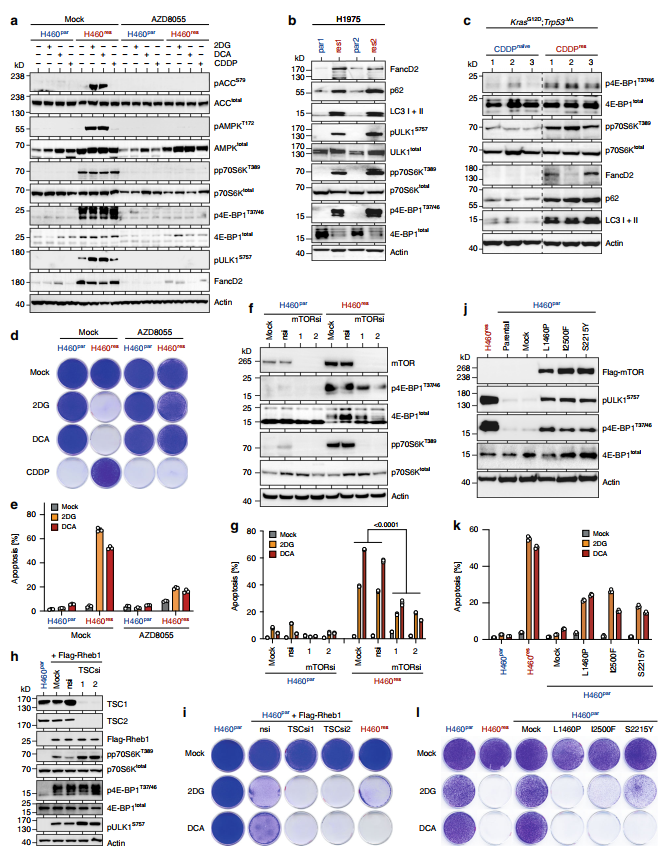
与此同时,耐CDDP的H460、H1975和小鼠腺癌细胞均显示mTOR信号通路明显升高,表现为mTORC1靶点磷酸化(p70S6KT389、4E-BP1T37/46和ULK1S757)和FancD2表达升高(图2a c)。
此外,ATP竞争性mTOR激酶抑制剂AZD805537和 mTORC1选择性抑制剂雷帕霉素和伊维莫司阻断了mTOR靶点的磷酸化,有效地使H460res细胞对CDDP重新敏感(图2a, d,补充图1)。
药物或rnai介导的mTORC1抑制使cddp耐药肿瘤细胞的药物应答模式恢复到亲代细胞的应答模式(图2d)。
mTOR抑制不仅恢复了H460res细胞对CDDP的敏感性,同时减弱了2DG/DCA的凋亡反应,挽救了在2DG/DCA存在下的克隆生长(图2d, e,补充图1和2)。
sirna介导的mTOR敲低以及mTORC1亚基Raptor敲低可以保护H460res细胞免受2DG和DCA的侵袭,而敲低mtorc2特异性亚基Rictor则没有影响(图2 f, g)。
综上所述,CDDP耐药肿瘤细胞中mTORC1信号通路的升高不仅是CDDP耐药表型所必需的,同时也是其代谢脆弱性的重要因素。
当内源性mTORC1激酶活性通过激活mtor GTPase Rheb1(脑内丰富的Ras同源物)的强化表达而提高时,H460par细胞对2DG/DCA变得敏感(图2 h, i)。
当rheb1 -抑制型TSC亚基被消耗后,TSC1和TSC2 mTORC1信号进一步增强,从磷酸化p70S6KT389、4EBP1T37/46和ULK1S757的水平可以明显看出,这导致了对2DG和DCA的更高敏感性(图2h, i)。
此外,在2DG/DCA存在的情况下,不同的mTOR突变体(之前在癌症患者12中发现,它们在H460par细胞中诱导了强mTORC1信号转导)对2DG/DCA诱导的细胞凋亡敏感,并导致克隆细胞生长受损(图2j l)。
因此,mTORC1信号的激活不仅是必需的,而且足以引起2DG和DCA的代谢脆弱性。

测量了氯喹阻断自噬溶酶体降解后LC3的积累(图3a)。与H460par细胞相比,H460res细胞中LC3的积累明显减少,表明在没有2DG/DCA的情况下,自噬通量明显降低。AZD8055抑制mTOR可以恢复H460res中LC3的积累,但在H460par细胞中没有明显的作用,说明H460res细胞基础自噬通量的降低是mTOR依赖性的。在2DG/DCA处理后,H460par细胞中LC3水平呈剂量依赖性下降,提示诱导自噬(图3b, c)。
与H460res细胞对2DG/ DCA处理后自噬反应的缺陷一致,只有H460par细胞在处理后出现大量的红色荧光LC3斑点(图3d, e)。
四、mTOR对2DG/DCA敏感,抑制自噬

各种药物自噬抑制剂作用于自噬过程的不同步骤,使H460par细胞对2DG/DCA敏感(图4a),支持了自噬在代谢紊乱背景下的保护作用。同样,必要自噬基因ATG7的下调使H460par细胞对2DG/ DCA敏感(图4b, c)。
由于药物抑制剂和siRNAs可能会产生脱靶效应和不完全抑制,我们还生成了多种由crispr诱导的ATG7插入/缺失突变的H460par细胞克隆(图4d f)。
只有完全敲除ATG7的克隆细胞显示自噬货物蛋白p62/SQSTM1的积累和LC3-I向LC3-II处理的失败,作为自噬缺陷的证实(图4d)。
在2DG/DCA处理下,这些克隆也显示了克隆细胞的生长和增殖受损(图4e, f)。
综上所述,这些实验明确地将自噬缺陷与2DG/DCA的抗肿瘤活性联系起来,并强调自噬缺陷是代谢药物易损的充分原因。在CDDP剂量增加的情况下,我们扩大了ATG-7修饰的H460par克隆,产生了具有不同ATG-7突变状态的耐CDDP的H460res克隆。
除ATG-7状态外,所有CDDP耐药克隆均对2DG/DCA过敏,但只有使用ATG-7的克隆通过mTOR抑制从2DG/DCA细胞毒性中拯救出来(图4g, h)。
五、mTOR介导的代谢脆弱性延伸到双胍类化合物
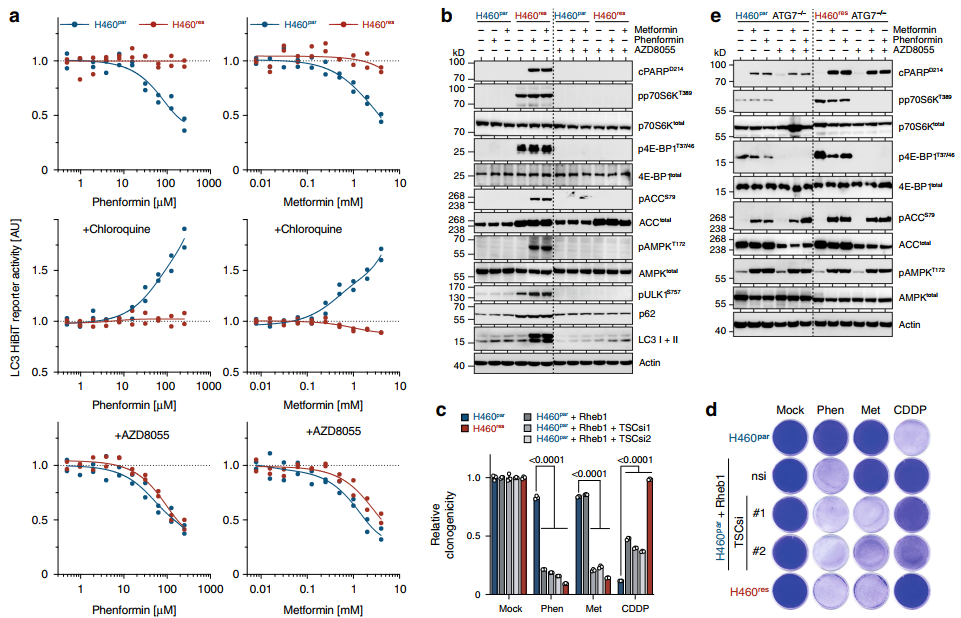
与2DG/DCA相似,H460par细胞通过诱导自噬通量对Met/Phen反应,在氯喹存在时LC3的剂量依赖性降解和积累证明了这一点(图5a)。
相比之下,H460res细胞证明只有微不足道的LC3的波动水平符合遇到的失败/苯酚的诱导自噬(图5)。
对Met/Phen处理的差异反应也与mTORC1有关。mTOR抑制恢复了Met/ pheno处理的H460res细胞中LC3的降解(图5a),显示了有效的自噬诱导,并挽救了这些细胞免于能量应激和凋亡(图5b)
相反,H460res细胞显示AMPKT172和ACCS79磷酸化和PARP裂解的增加,分别作为能量应激和随后发生的细胞凋亡的信号(图5b,左图),以及Met/Phen抑制克隆生长的信号(图5d)。
由于Rheb1表达和TSC缺失,mTORC1信号强烈地降低了Met/Phen处理的H460par细胞的克隆生长(图5c, d)。因此,提高mTORC1信号是满足Met/Phen敏感性的必要条件。
六、代谢脆弱性与靶向耐药性相关

我们生成T47D乳腺癌细胞的alpelisib和pictilisib耐药亚克隆,显示交叉耐药(图6a)。
p70S6KT389、4E-BP1T37/46和ULK1S757磷酸化水平的降低证明了两种药物都抑制了亲代细胞中的mTORC1信号传导(图6b)。
与之前的报道一致,PI3Ki耐药细胞在PI3K抑制下维持mTORC1信号通路,这是耐药的根本原因(图6b)。
mTORC1信号通路抑制了自噬,抗pi3ki的T47D克隆在2DG/DCA处理后未能诱导自噬通量(图6c)。
值得注意的是,抑制PI3K也刺激了LC3处理和p62降解,表明自噬诱导再次只在亲本亚克隆,而不耐PI3Kiresistant亚克隆(图6b)。
mTOR抑制后,2DG/DCA恢复了自噬诱导,表明耐PI3Kiresistant肿瘤细胞自噬反应存在严重的mTOR依赖性缺陷。与我们之前的结果一致,这转化为通过2DG/DCA治疗,mTOR依赖大量诱导细胞凋亡和降低克隆生长(图6d, e),这将我们对mTOR诱导代谢脆弱性的研究从传统化疗扩展到靶向治疗耐药性。
七、代谢抑制剂在体内靶向CDDP耐药的癌细胞
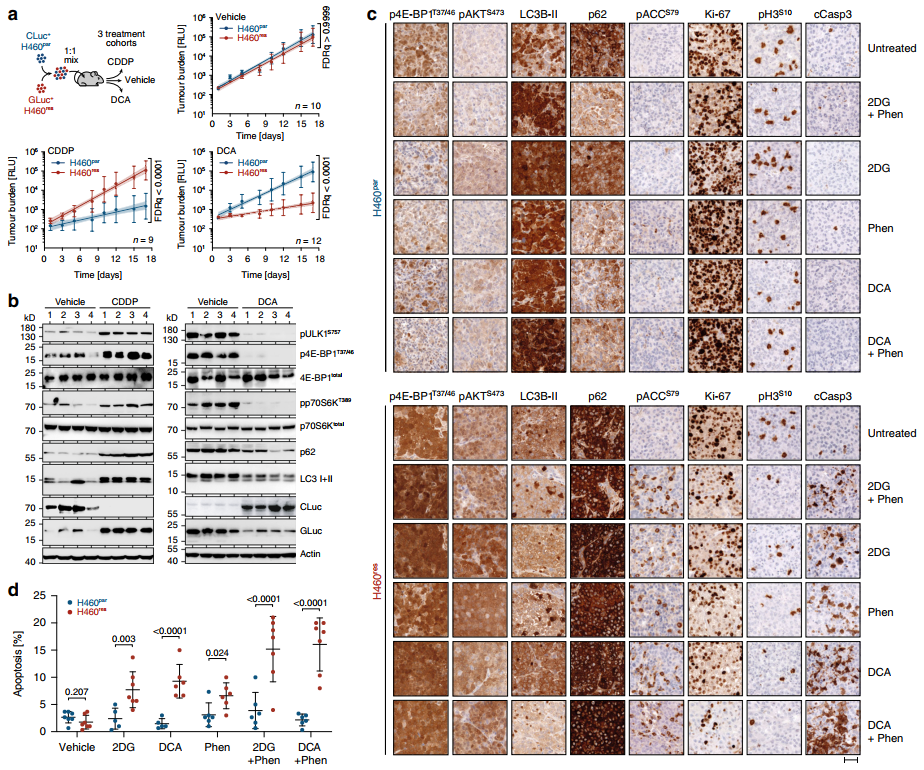
治疗期间定期采集的血液样本的荧光素酶活性测量提供了治疗期间肿瘤负荷的细胞类型特异性纵向监测(图7a).
DCA选择性地抑制H460res细胞的生长,导致终末期肿瘤中H460res细胞减少40倍。为了证实观察到的细胞丰度变化,我们分析了终末期小鼠的肿瘤裂解液中mTOR和自噬标记物(图7b)。
与由两种肿瘤细胞类型组成的未治疗小鼠的肿瘤相比,cddp治疗组的肿瘤表现出mTOR信号上调、LC3处理缺陷和p62降解(图7b,左),这与基于荧光肿瘤监测观察到的cddp诱导的H460res细胞富集相一致。相比之下,DCA处理的小鼠肿瘤显示减少的mTOR信号和减少的LC3水平和p62(图7 b,右面板),符合DCA H460res细胞耗竭。
在H460par肿瘤中,mTOR信号在治疗期间保持较低或下降,而在H460res肿瘤中,mTOR信号维持甚至上升(图7c)。
最重要的是,在H460res肿瘤中,作为细胞凋亡标志物的cleaved - caspase-3在单次2DG或DCA处理后显著上调(图7c, d,补充图6)。
单独使用Phen时,虽然2DG/DCA的剂量降低到50%,但仅轻微增加了细胞凋亡,但却大大增强了2DG/DCA的凋亡作用(图7d)。