






泛素化酶VS巨噬细胞极化
导语:巨噬细胞是先天性免疫系统的效应细胞,对宿主防御、炎症消退和伤口愈合至关重要。巨噬细胞有促炎性和抗炎性的两个极端。了解调控巨噬细胞极化的机制,对于宿主防御和修复组织至关重要。泛素特异性蛋白酶19(USP19)是一种内质网(ER)锚定的去泛素化酶,调节ER相关蛋白降解和DNA损伤修复。泛素化酶与巨噬细胞极化的研究值得关注。

(IF=8.484)
技术路线

结果
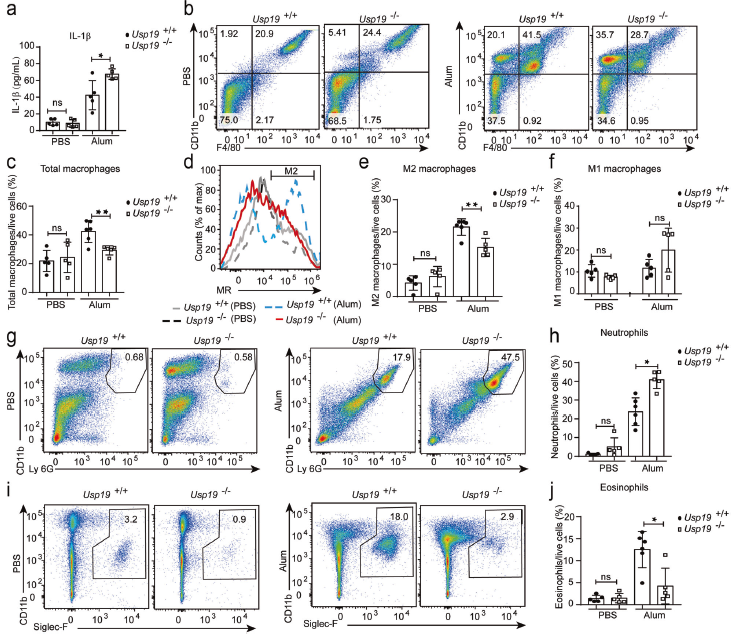
为揭示USP19在炎症中的作用,使用铝盐(明矾)诱导的腹膜炎模型来检测IL-1β的产生,发现Usp19-/-小鼠的灌洗液中IL-1β的浓度显著高于野生型小鼠。为确定Usp19-/-小鼠IL-1β产生的增加是否与巨噬细胞募集的增加有关,定量灌洗液中的巨噬细胞。发现明矾处理后Usp19-/-小鼠中巨噬细胞总数减少。因此,推断Usp19-/-小鼠M1样巨噬细胞和M2样巨噬细胞之间的平衡可能发生改变。明矾处理后,Usp19-/-小鼠中M2样巨噬细胞(F4/80 + CD11b + MR +)而非M1样巨噬细胞(F4/80 + CD11b + CD86 +)的比例显著降低。巨噬细胞极化与中性粒细胞和嗜酸性粒细胞募集密切相关,发现Usp19-/-小鼠中明矾诱导的中性粒细胞募集增加,嗜酸性粒细胞募集严重受损。这些结果证明Usp19缺陷在明矾诱导的腹膜炎模型中将抗炎反应转换为促炎反应。
2. USP19可促进自噬介导的活性氧(ROS)清除,抑制NLRP3炎症小体的激活
鉴于USP19在明矾诱导的腹膜炎模型中显著抑制IL-1β的产生,推测USP19可能调节炎症小体的激活。从Usp19-/-小鼠中分离出不同的细胞类型,发现在骨髓源性巨噬细胞(BMDMs)、骨髓源性树突状细胞(BMDC)和小鼠胚胎成纤维细胞(MEFs)中,Usp19缺失显著增加ATP、明矾和二氧化硅刺激引起的IL-1β释放,而AIM2炎症小体激活剂诱导的IL-1β分泌保持不变。ATP处理的Usp19-/-小鼠BMDMs上清液中裂解的caspase-1增加。炎症小体激活过程中,NLRP3通过PYD–PYD介导的相互作用触发ASC斑点的形成。在USP19-KO THP-1细胞中,脂多糖LPS致敏和ATP激发的THP-1来源的巨噬细胞中ASC斑点形成在炎症小体激活后显著增强,在USP19-KO THP-1来源的巨噬细胞中敲低内源性NLRP3,发现NLRP3的耗竭消除了USP19-KO细胞对LPS和ATP的IL-1β上调反应。这些表明USP19缺陷特异性地增强NLRP3炎症小体激活。
为确定USP19是否通过ROS的产生影响NLRP3炎症小体的激活,检测发现USP19缺乏增强了ROS的产生,线粒体靶向抗氧化剂处理巨噬细胞可消除Usp19-/-巨噬细胞中IL-1β分泌的增加。这些表明,USP19的耗竭通过上调线粒体ROS产生增强NLRP3炎症小体激活。在缺乏Beclin-1的情况下,Usp19缺失导致的ROS上调降低,Beclin-1的缺失消除了USP19对IL-1β分泌的影响。这些表明,USP19/Beclin-1的缺失损害了自噬,导致ROS产生增加和随后的NLRP3炎症小体激活。

3. USP19对M2样巨噬细胞极化至关重要
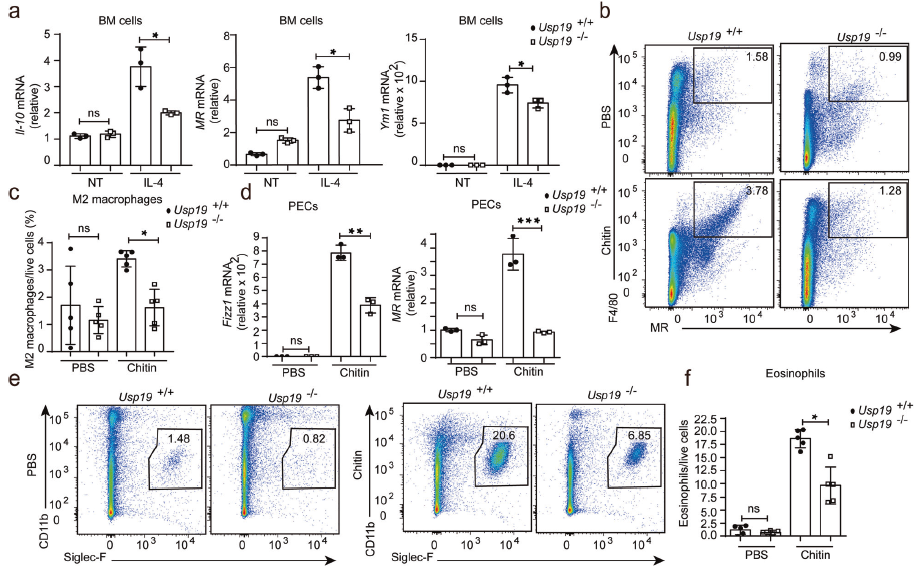
IL-4处理WT和Usp19-/-巨噬细胞,发现M2样巨噬细胞相关标记物显著降低。给小鼠腹腔注射甲壳素,发现从Usp19-/-小鼠获得的腹腔中M2样巨噬细胞比例降低,嗜酸性粒细胞数量也减少。这些表明USP19对甲壳素给药后促进M2样巨噬细胞极化至关重要。
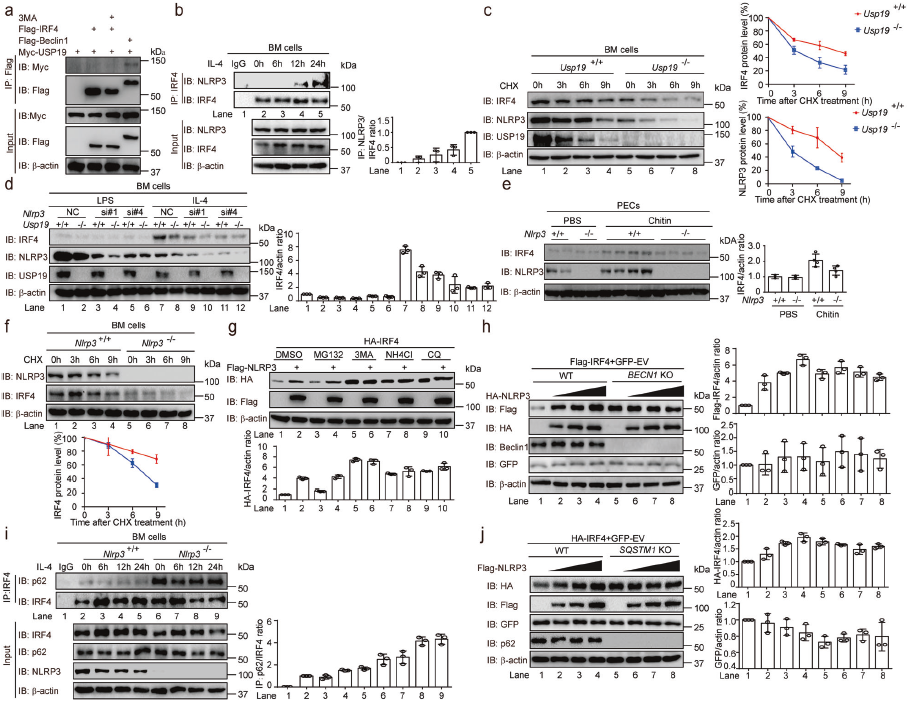
4. USP19阻止IRF4被p62介导的选择性自噬降解
放线菌酮试验发现USP19缺陷导致M2样巨噬细胞的关键转录因子IRF4显著降解,USP19的过表达增加了NLRP3稳定表达的人胚肾细胞中IRF4蛋白水平。自噬抑制剂、溶酶体抑制剂以及蛋白酶体抑制剂可完全抑制USP19介导的IRF4稳定作用。检测IRF4与不同受体的相互作用,发现IRF4与p62特异性相互作用,USP19缺陷显著增强p62和IRF4之间的关联。在Usp19-/-BM细胞中重新表达了小鼠IRF4,恢复了IL-4诱导的Usp19-/-BM细胞中M2样巨噬细胞极化。这些表明USP19通过阻止IRF4被p62介导的选择性自噬降解,促进M2样巨噬细胞极化。

5. USP19通过NLRP3稳定IRF4
为研究USP19如何阻止p62介导的IRF4降解。检测到NLRP3缺陷降低了M2样巨噬细胞的比例,NLRP3-/-BM细胞中小鼠NLRP3的再表达恢复IL-4诱导的M2样巨噬细胞极化。IL-4刺激的BM细胞中,IRF4与NLRP3相关,并且IRF4的N末端对于IRF4–NLRP3结合至关重要。Usp19缺陷通过影响NLRP3和IRF4的降解速率而降低其蛋白水平。敲除NLRP3可显著降低USP19诱导的IRF4蓄积。溶酶体抑制剂单独稳定IRF4,加入NLRP3并不能进一步稳定IRF4。在自噬严重受损的BECN1-KO细胞中,NLRP3不能进一步增加IRF4蛋白水平。外源性NLRP3消除了IRF4–p62相互作用,NLRP3缺陷显著增加了IL-4诱导的IRF4–p62结合。这些表明NLRP3通过阻断IRF4–p62相互作用抑制IRF4降解。
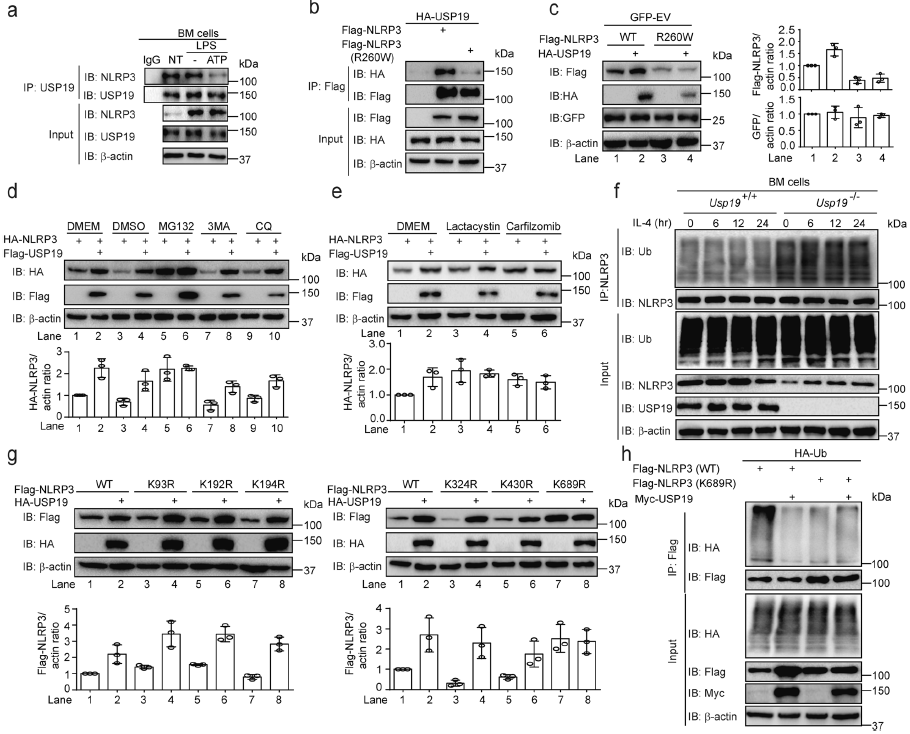
6. USP19与NLRP3相互作用并稳定NLRP3
在BM细胞中,内源性USP19和NLRP3直接相关,但其相互作用因炎症小体激活而降低。NLRP3 R260W突变体是NLRP3的一种活性形式,在没有刺激的情况下直接触发IL-1β分泌,结合USP19的能力下降,USP19不能稳定NLRP3 R260W突变体。在蛋白酶体抑制剂存在的情况下,USP19介导的NLRP3蓄积消失。USP19在K689切割NLRP3的多聚泛素链,抑制NLRP3的蛋白酶体降解。
总结:
1. USP19促进自噬,清除ROS,下调NLRP3炎症小体激活。
2. USP19通过去除NLRP3的K689上的多聚泛素链,稳定炎症小体中未掺入的NLRP3,促进NLRP3与IRF4的相互作用,从而阻止IRF4的p62依赖的选择性自噬降解。
