抑制自噬增强了Nrf2促进小鼠糖尿病心肌炎
Nrf2能够改善或加重糖尿病心肌病,但是其潜在的机制却没有报道。Huimei Zang的等人在Diabetes(IF=7.72)这一杂志中......
研究背景:
Nrf2能够改善或加重糖尿病心肌病,但是其潜在的机制却没有报道。Huimei Zang的等人在Diabetes(IF=7.72)这一杂志中发表文章,介绍Nrf2介导的1型糖尿病心肌损伤的新机制。
技术路线图:
研究结果:
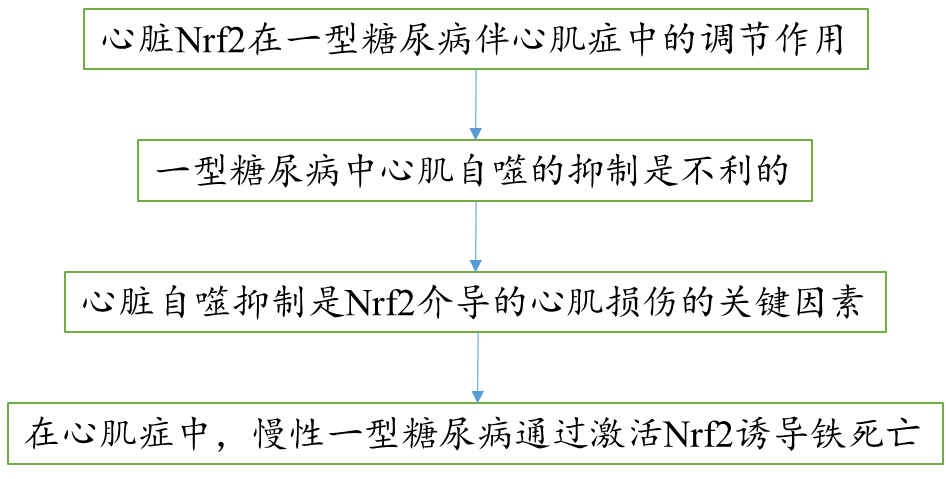
1.心脏Nrf2在一型糖尿病伴心肌症中的调节作用
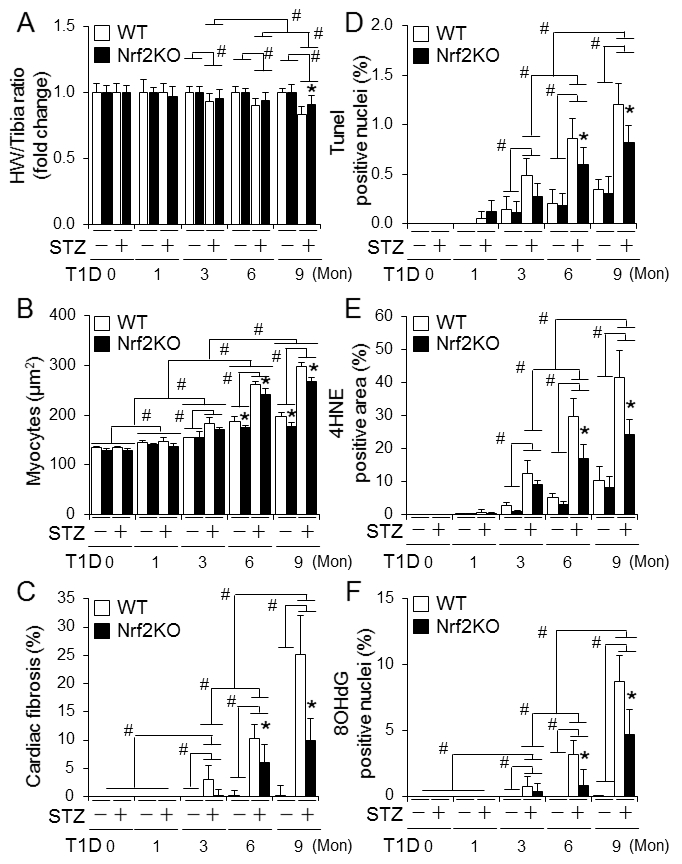
Nrf2敲除的小鼠中糖尿病相关死亡增加,而Nrf2KO改善空腹血糖和糖尿病相关体重的减少(Fig. 1A)。糖尿病发病6个月,Nrf2KO不影响心肌症紊乱的发生,但是减轻9月的病情进展(Fig. 1C)。另外,心肌肥大,细胞死亡,纤维化和氧化应激在中期和早期阶段能够被Nrf2KO所缓解(Fig. 2)。Nrf2KO小鼠2月-11月中出现病理性心脏生长比WT小鼠中较轻。为了避免其他因素干扰,比较了FS,BM和心脏重量/胫骨比例的变化。总的来说,Nrf2对一型糖尿病和相关死亡起到总体保护作用。但是它对糖尿病伴有心肌症的发展并没有保护或者延迟作用。相反,它可能以性别非依赖方式促进疾病的发展。在分子水平上,Nrf2可能增强糖尿病心脏的氧化压力,从而促进糖尿病心肌症的发展。
Fig.1
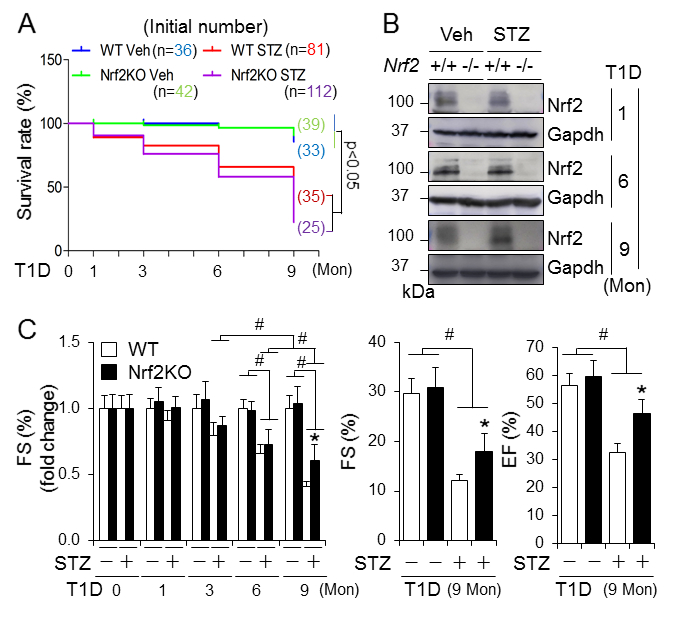
Fig.2
2.一型糖尿病中心肌自噬的抑制是不利的
CR-Atg5KO诱导心肌自噬抑制。心肌自噬流在3个月时是完整的,但是在糖尿病发病6个月时显著抑制,暗示在一型糖尿病中,高脂血症在抑制心肌自噬中的分担作用。接下来,在表达mCherry-Gfp-Lc3受体的H9C2心肌细胞样细胞中检测自噬流和自吞噬泡流出。因自噬溶酶体被红色荧光标记,自噬体被黄色荧光标记(红色和绿色)。H9C2细胞中基础自噬以自噬溶酶体居多和少量自噬体为主(Fig.3B)。表明大多数合成的自噬体不断与溶酶体融合形成自噬溶酶体,并进行降解。因此,溶酶体抑制剂BafA1用于控制自噬,来区分自噬体与溶酶体融合的数量。在H9C2细胞中,棕榈酸盐负担抑制了自噬特征通过损害自噬溶酶体流,并且在高糖环境中,脂质过量诱导的自噬抑制加重(Fig. 3B)。然而,CRAtg5KO导致心肌紊乱的早期发生,并随着时间的推移促进了它的发展(Fig.4A, 4C)。CRAtg5KO同样加速了糖尿病诱导的心肌病例改变,包括心肌纤维化,心肌肥大,细胞死亡和氧化应激(Fig. 4B-C)。另外,Atg5的敲除增强了H9C2细胞死亡,但是在脂质积累伴有高糖环境中没有改变,暗示抑制自噬是心肌细胞脂肪毒性和/或葡萄糖毒性的参与机制。结果表明随着时间进展,糖尿病通过抑制心肌自噬,诱导葡萄糖依赖的心肌细胞死亡,从而导致糖尿病心肌病的发病机制
Fig.3
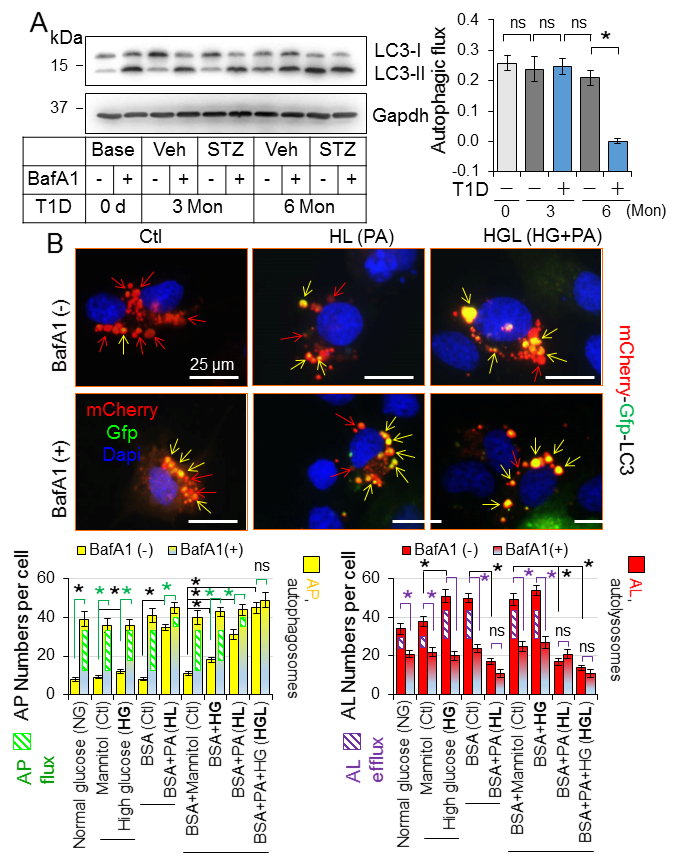
Fig.4
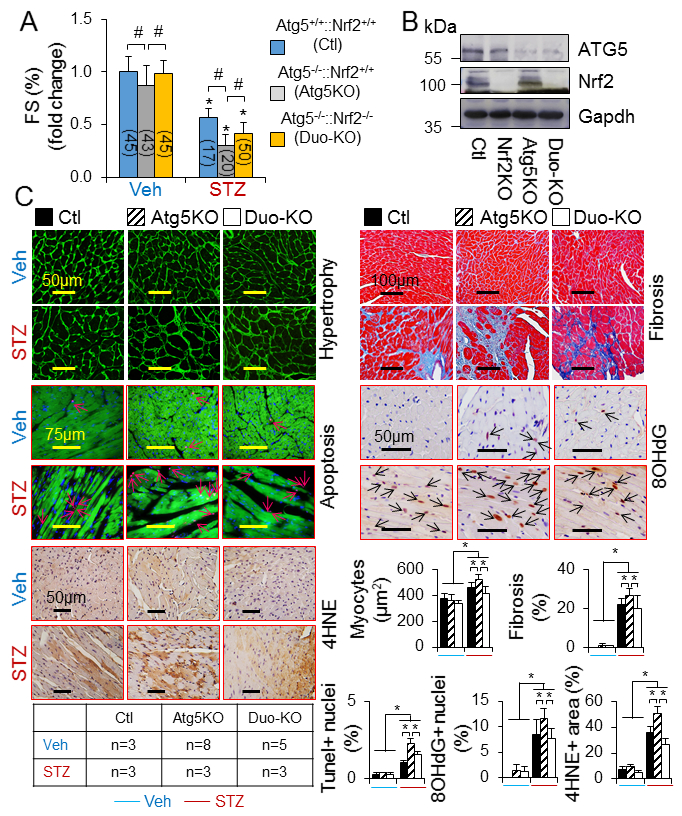
3. 心脏自噬抑制是Nrf2介导的心肌损伤的关键因素
之后,我们研究了心肌自噬抑制对驱动Nrf2介导的心肌损伤的作用,并且是否能促进心肌症相关一型糖尿病的进程。选择糖尿病发病9个月的实验组,CR-Atg5KO增强了糖尿病诱导的心脏病理重塑和功能障碍(Fig. 5)。这些不良的表型被Nrf2KO所缓解。因为CR-Atg5KO本身不会影响糖尿病的状态。这些结果暗示自噬抑制-Nrf2激活-糖尿病中的心肌损伤和功能紊乱。
Fig.5
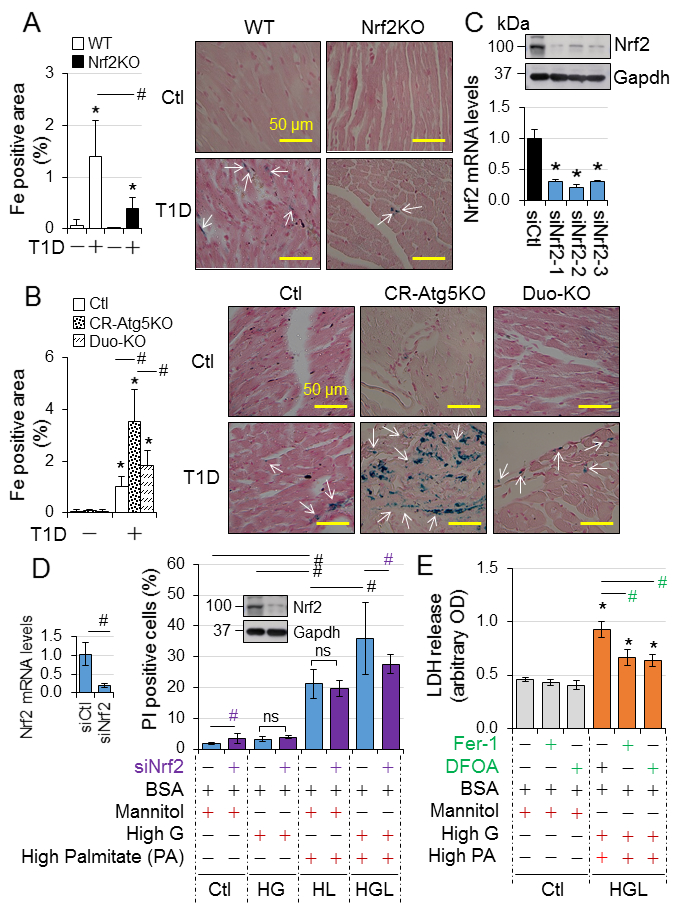
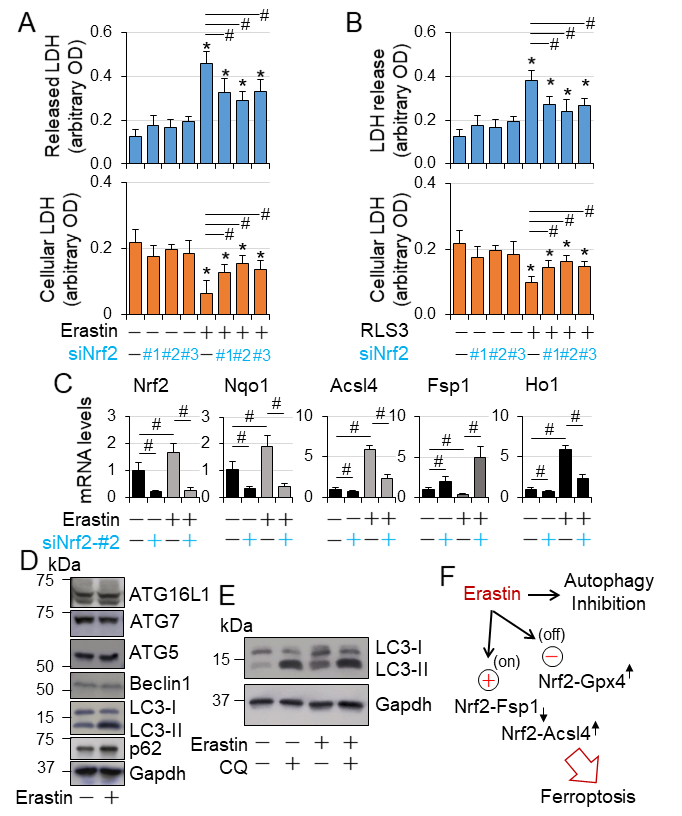
研究显示Nrf2在阿霉素诱导的铁死亡中可能有调节作用。在心肌细胞中,脂质过氧化物致死积累而产生的铁依赖形式的细胞死亡,通过上调心肌细胞中HO-1的表达。在糖尿病发生9月份,心肌铁沉积增加;CR-Atg5KO糖尿病诱导的铁沉积增强;然而NRF2敲除缓解了这两种增加(Fig. 6A-B)。Fig. 2E,5C显示铁死亡脂质过氧化的生物标记物与4HENE在糖尿病中的改变显示出相似的模式。这是Nrf2通过增强一型糖尿病铁沉积和脂质过氧化,促进心肌细胞铁死亡。敲除Nrf2选择性抑制高脂毒性在铁死亡抑制剂,Fer-1,铁螯合剂去铁胺(DFO)抑制糖脂毒性在H9C2细胞中(Fig. 6C-E),提示一型糖尿病晚期发生的糖醛酸中毒驱动了nrf2介导的心肌细胞铁死亡。结果提示心肌细胞中存在慢性一型糖尿病-自噬抑制- nrf2激活-铁死亡的信号通路。
然后,我们发现在H9C2细胞中Nrf2敲除对铁死亡的作用。在保持自噬完整性的正常葡萄糖水平培养条件下,敲除Nrf2增加细胞死亡(图7A-B)。表明Nrf2对自噬功能正常的心肌细胞具有细胞保护作用。同样,敲除Nrf2(使Nrf2失活)抑制基础的和erastin诱导的Acsl4的表达,但是逆转了erastin诱导的Fsp1表达的抑制当上调基础表达(Fig. 7C)。这些结果显示Nrf2在抑制心肌细胞Fsp1的同时促进Acsl4的表达,在GPX4失活的情况下这种作用可能会加剧导致铁死亡。
一型糖尿病诱导的自噬抑制可能激活nrf2介导的心肌细胞铁死亡,从而促进糖尿病心肌病的进展。
Fig.6
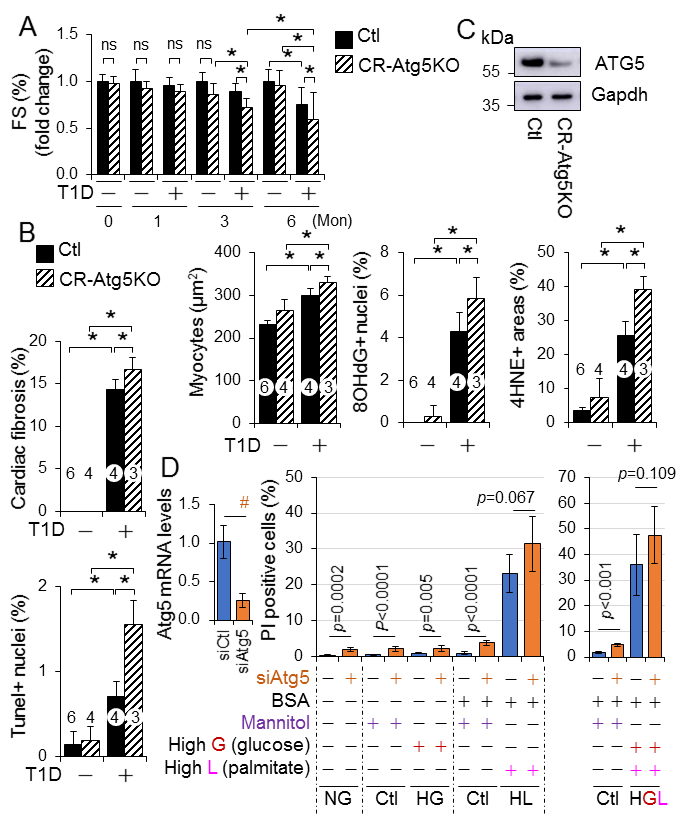
Fig.7