






支链氨基酸转氨酶2在癌细胞中调节铁死亡
2020年10月江苏大学附属医院Haitao Zhu教授于Cell Death & Differentiation上发表文章Branched-chain amino acid aminotransferase 2 regulates ferroptotic cell death in cancer cells其研究发现确认了BCAT2在铁死亡中的新作用,提示了一种克服索拉非尼耐药的潜在治疗策略。

铁死亡是一种由细胞氧化还原稳态丧失引起的细胞凋亡,导致脂质过氧化失控,最终导致细胞死亡。胱氨酸-谷氨酸抗氧化剂(系统Xc)或谷胱甘肽过氧化物酶4 (GPX4)的药理失活可诱导铁死亡增多,提示谷胱甘肽依赖的抗氧化防御在防止铁死亡细胞死亡中起关键作用。铁死亡作用与缺血引起的器官损伤、与退行性疾病相关的病理细胞死亡以及不同类型的癌症有关。多种肿瘤细胞易发生铁死亡增多,包括淋巴瘤、宫颈癌、头颈癌、胰腺癌、肾细胞癌和肝细胞癌。各种研究证实了铁亡诱导剂的关键作用,包括小分子铁亡诱导剂如伊拉斯汀,以及许多药物(如索拉非尼、青蒿素及其衍生物)在杀伤肿瘤细胞和抑制肿瘤方面的作用增长。这些铁死亡诱导剂还可与化疗药物协同治疗肿瘤。有趣的是,有些类型的癌症对铁死亡诱导剂比其他类型的更敏感。反向过硫通路已被确定为铁死亡作用的负调节因子,卵巢癌细胞中缺乏该通路与伊拉斯汀诱导的铁死亡敏感性增高有关。HSF1-HSPB1通路也在人宫颈癌、前列腺癌和骨肉瘤中负调控星抑素诱导的铁死亡作用。MUC1-C/xCT通路是伊拉斯汀诱导的三阴性乳腺癌细胞铁胞作用的另一个负调控因子。
越来越多的证据表明,细胞代谢在铁死亡中起着至关重要的作用[12,13]。转录因子NRF2协调抗氧化防御系统调节铁死亡作用。p62- Keap1-NRF2是[14]肝癌细胞上铁死亡的中枢抑制通路。基因或药物抑制NRF2显著增强铁死亡 伊拉斯汀引起的肝癌易感性和索拉非尼,而NRF2表达式led的激活细胞抵抗铁死亡,建议中央部分含氧降低分子的作用,特别是活性氧(ROS)在铁死亡。细胞内的铁代谢对铁依赖氧化酶或芬顿化学作用也是必不可少的。最近的一项研究表明,铁死亡的自噬降解通过自噬货物受体核受体共激活因子4 (NCOA4)调节铁死亡的作用。不足为奇的是,氨基酸代谢也参与了铁死亡。高浓度的胞外谷氨酸,伊拉斯汀,或其他系统Xc抑制剂阻断胞内胱氨酸/半胱氨酸摄取,以诱导铁死亡。沉默cysteinyltRNA合成酶(CARS)可上调经硫途径,从而导致对伊拉斯汀诱导的铁死亡的抗性。谷氨酰胺通过其特定的代谢酶谷氨酰胺酶(GLS1和GLS2)介导铁死亡作用,但谷氨酰胺分解过程的机制复杂。然而,在肝癌细胞中控制铁死亡敏感性的代谢途径尚不清楚。在本研究中,作者鉴定了支链氨基酸氨基转移酶2 (BCAT2),一种氨基转移酶催化硫氨基酸代谢,作为铁死亡的特异性抑制剂。作者发现BCAT2参与了Xc系统抑制物诱导的肝癌细胞铁死亡作用。此外,BCAT2还参与了磺胺嘧啶与索拉非尼协同诱导铁死亡的机制。因此,作者的研究结果表明BCAT2是铁死亡的抑制因子,并参与了肝癌铁死亡的核心代谢信号通路。
在这项研究中,作者证实了伊拉斯汀、索拉非尼或磺胺嘧啶激活了铁死亡,增加了细胞不稳定铁的水平。细胞内高水平的不稳定铁导致细胞内ROS的快速积累,这是铁中毒的必要条件。有趣的是,作者发现该噬铁途径也激活AMPK磷酸化,从而抑制SREBP1的核易位,并抑制其直接靶基因BCAT2的转录(图5)。作者进一步发现,BCAT2通过调节细胞内谷氨酸水平抑制了细胞内的铁死亡。重要的是,在体外和一些动物模型中,如皮下胰腺癌模型、原位肝癌模型和PDX肝癌模型中,硫柳氮嗪与索拉非尼的联合在抑制BCAT2表达和促进铁毒性癌细胞死亡方面具有协同作用。重要的是,在这些临床前癌症模型中,BCAT2也显示出作为评估药物反应的敏感生物标志物的潜力。BCAT2控制铁死亡的发现与氨基酸在[21]铁死亡中起关键作用的概念一致。BCAT是关键的代谢蛋白,催化BCAA的可逆转氨基反应生成各自的a-酮酸(BCKAs)和谷氨酸,负责生成30%的从头脑谷氨酸。谷氨酸的代谢与铁死亡的调控密切相关。值得注意的是,Xc系统的功能是由谷氨酸水平调节的,因为在Xc系统中谷氨酸以1:1的比例交换胱氨酸。因此,细胞外高浓度的谷氨酸阻断系统Xc活性,抑制胱氨酸摄取,并驱动铁死亡。相比之下,作者体外实验中细胞内高水平的谷氨酸来源于BCAT2驱动的谷氨酸的重新合成,从而可能增强系统Xc活性,促进胱氨酸摄取,抑制铁亡。BCAT2细胞内谷氨酸代谢的这种保护作用与细胞外脑谷氨酸水平降低对Xc敲除小鼠神经毒性损伤的保护作用是一致的。此外,作者的实验证实了BCAT2的表达水平受到AMPK-SREBP1信号通路的调控,这也得到了之前在胰腺癌[18]中BCAT2基因组缺失导致侧死的发现的支持。作为代谢应激的关键传感器,AMPK在铁死亡作用中的作用与环境有关。AMPK通过介导BECN1磷酸化和BECN1- slc7a11复合物的形成,从而促进癌细胞的铁死亡,这与作者发现的铁死亡抑制剂通过激活AMPK而下调BCAT2表达的研究结果一致。有趣的是,作者注意到最近的报道,能量应力介导的AMPK激活抑制了铁死亡作用。AMPK在铁死亡中作用的差异可能与被测细胞系的基础AMPK活性和环境背景有关。因此,AMPK在癌症铁死亡中的潜在作用值得深入研究,也为肿瘤患者提供了一种重要的治疗策略。
BCAA是合成谷氨酸和谷氨酰胺的氮供体,但谷氨酰胺在铁死亡中的作用是复杂的。谷氨酰胺是通过其特殊的代谢-谷氨酰胺分解来降解的。当glutaminolysis抑制或谷氨酰胺耗尽,胱氨酸饥饿和阻断胱氨酸进口均不能诱导ROS积累、脂质过氧化和铁死亡增多,说明谷氨酰胺溶解是铁死亡增多的燃料。根据这一观察,除谷氨酰胺外,谷氨酰胺分解的另一产物a-酮戊二酸(a-KG)可替代谷氨酰胺诱导铁死亡。作者推测BCAT催化BCAAs-BCKAs穿梭合成谷氨酸,导致细胞内a- kg水平降低,这可能是引起铁死亡的另一个原因。敲除BCAT2对SLC7A11的表达无影响。因此,BCAT2与系统Xc之间的确切关系需要在今后的工作中进行更深入的研究。肝癌是全球癌症死亡的第三大原因,而标准的化疗对大多数肝癌患者并没有效果,医生一直在寻找靶向疗法。索拉非尼是唯一的多激酶抑制剂作为一线治疗被证明可以延长不可切除的HCC[26]的总生存率。然而,来自亚太地区服用索拉非尼的患者的总生存率仅为6.5个月,反应率低为2%。
最近,最近的一项三期临床研究中,其总体生存优势并没有超过索拉非尼。在这项研究中,作者发现磺胺嘧啶单独或与索拉非尼联合,在二铁诱导疗法中发挥作用。这些发现是相应的,因为磺胺嘧啶是一种抗炎药物,已经广泛用于炎症性肠病的慢性、长期治疗,保证了其在成人和儿童中的安全性。基于作者的研究结果,磺胺嘧啶可能是晚期肝癌以及其他无法切除的癌症的一种潜在的新治疗选择。由于治疗中表达的变化,BCAT2可能是最敏感的靶点之一,其表达可作为预测索拉非尼和磺胺嘧啶联合治疗反应的标志物。然而,这一假设应该在患者资料中进行评估。综上所述,作者的数据表明,抑制细胞内谷氨酸的合成可以作为在癌症背景下诱导铁死亡依赖性的一个好策略。作者的研究结果支持了这一观点,即硫磺胺嘧啶与索拉非尼协同下调BCAT2,从而下调细胞内的谷氨酸。作者的工作还表明,细胞杀伤机制涉及调控从头合成谷氨酸是肝癌细胞的关键过程。作者认为BCAT2的蛋白或mRNA水平可用于预测癌细胞对未来铁运诱导疗法的反应性。作者还提出,高特异性的BCAT2抑制剂可以为一部分有意义的癌症患者提供有效的治疗。
技术路线:
一、用kinome CRISPRa筛选鉴定铁死亡的新作用以及Ferroptosis诱导物通过ferritinophagy-AMPK-SREBP1通路抑制BCAT2的表达
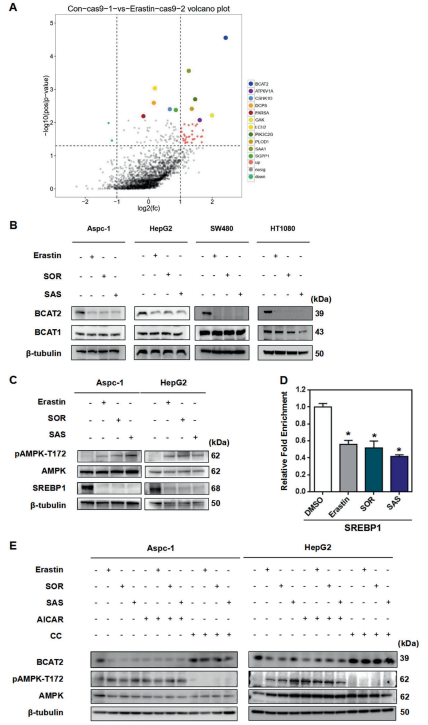
A. 为了系统阐明ferroptosis下游保守负调控因子,我们进行了大规模的基因CRISPR激活(CRISPRa)筛选。靶向2320激酶、磷酸酶和药物靶点(https://www.addgene.org/ pooled-library/weissman-human CRISPRa -v2-subpools/)的pooled人CRISPRa sgRNA慢病毒库和Cas9-VPR酶通过慢病毒转导引入HepG2细胞,然后用erastin处理或对照DMSO处理.富集sgRNAs的基因靶点是在HepG2细胞中赋予erastin介导的ferroptosis抗性的潜在基因。在我们的筛选方法中,许多报道的ferroptosis基因被鉴定和验证。其中分支的BCAT2是ferroptosis潜在负调控因子的首选基因.
B. 在ferroptosis诱导剂处理后,BCAT2的蛋白和mrna表达水平有所下降,但BCAT1 (BCAT2的同源物)没有下降,
在甲磺酸二铁胺(DFO,一种ferroptosis抑制剂)的存在下被逆转。
C. 推测ferroptosis诱变剂通过AMPK-SREBP1信号通路下调BCAT2。通过western blotting定量分析,erastin、sorafenib或s ulfasalazine诱导了苏氨酸残基172 (T172)上AMPK磷酸化,并降低了SREBP1的表达(图1C、S4A和S4B)。
D. ChIP实验还显示,erastin、sorafenib或sulfasalazine存在时,SREBP1与BCAT2的结合显著减少.
E. AMPK激活剂AICAR下调Aspc-1和HepG2癌细胞中BCAT2的表达方式与erastin、sorafenib或sulfasalazine类似,AMPK抑制剂化合物C可逆转这一过程,进一步证实了ferroptosis诱变剂通过激活AMPK下调BCAT2的表达.
二、BCAT2是癌细胞铁死亡的抑制因子

A.考虑到铁在铁死亡中的关键作用,我们首先研究了BCAT2表达与铁积累的相关性。
与预期的一样,erastin、sorafenib或sulfasalazine处理在对照组和BCAT2转染的细胞中都诱导了游离铁的积累.
B. erastin, sorafenib, and sulfasalazine 处理后,Aspc-1和HepG2细胞的GSH水平受到抑制,BCAT2的异位表达恢复了GSH水平.
C.与亲代细胞相比,erastin, sorafenib, and sulfasalazine处理后,脂质过氧化的终产物丙二醛(MDA)水平在Aspc- 1和HepG2细胞中升高,而过表达bcat2的细胞中降低(图2C和S8D)。
D-F.与这些结果一致,在erastin, sorafenib, and sulfasalazine存在下,BCAT2过表达增加了细胞内谷氨酸(图2D和S8E)和谷氨酸释放(图2E和S8F),并以时间依赖的方式减少了 system Xc抑制剂诱导的细胞死亡(图2F和S8G).
G-L.在erastin, sorafenib, and sulfasalazine存在的情况下,敲除BCAT2-显著增加了Aspc-1和HepG2细胞中MDA的生成(图2H和S11D)和GSH的消耗(图2I和S11E),但对游离细胞铁的积累没有影响(图2G和S11C)。此外,BCAT2在erastin, sorafenib, and sulfasalazine存在下,下调细胞内谷氨酸水平(图2J和S11F),谷氨酸释放(图2K和S11G)以及细胞活力(图2L和S11H)。用RSL3和BSO处理亲代和BCAT2敲除Aspc-1和HepG2细胞,显示出相似的结果(图S12)。因此,BCAT2沉默细胞的集落形成能力受到抑制(图S11I)。敲除BCAT2并不影响Aspc-1和HepG2细胞中SLC7A11和GPX4蛋白的表达水平(图S13A)。这些结果表明,BCAT2敲低可部分诱导铁死亡。
三、BCAT2参与sulfasalazine和 sorafenib协同诱导铁死亡的机制
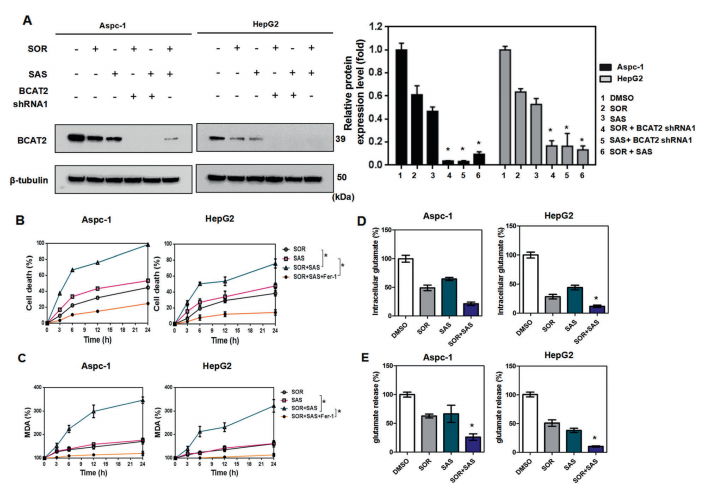
A-E.sorafenib and sulfasalazine联合应用可显著抑制Aspc-1和HepG2细胞中BCAT2的表达(图3A),与sorafenib and sulfasalazine单独联合BCAT2 shRNA的模式相似(图2M和S11J)。sorafenib and sulfasalazine在增加细胞死亡(图3B)和MDA生成方面也表现出协同作用,在ferrostatin-1 存在时可以恢复这一作用(图3C),抑制细胞内谷氨酸水平(图3D),谷氨酸释放(图3E)。
总之,sorafenib and sulfasalazine通过部分调节BCAT2表达对ferroptosis的影响。

在C57BL/6小鼠中,在C57BL/6小鼠中,sorafenib and sulfasalazine分别减少了9.63%和13.5%的Panc02皮下肿瘤大小,联合治疗在第14天进一步减少了81.39%的肿瘤大小(图4A-C)。
由于原位异种移植模型在复制肿瘤微环境和预测药物疗效方面被认为优于皮下肿瘤模型,
在C57BL/6小鼠H22细胞的原位HCC模型中,我们想验证 sulfasalazine诱导铁死亡是否也能增强索拉非尼的抗癌活性(图4D)。
sorafenib and sulfasalazine显著减小了原位异种移植瘤的肿瘤大小(图4E),延长了动物存活时间(图4F)。
此外,sorafenib and sulfasalazine联合治疗显著降低了蛋白和mrna表达水平中的BCAT2(图s)。S14A和S14B),
原位HCC组织中GSH水平降低(图S14C), MDA水平升高(图S14D)。在C57BL/6小鼠H22细胞的原位HCC模型中,我们想验证磺胺吡啶诱导二茂铁tosis是否也能增强索拉非尼的抗癌活性(图4D)。的确,索拉非尼联合柳氮磺胺吡啶显著减小了原位异种移植瘤的肿瘤大小(图4E),延长了动物存活时间(图4F)。
此外,索拉非尼和柳氮磺胺嘧啶联合治疗显著降低了蛋白和mrna表达水平中的BCAT2(图s)。S14A和S14B),原位HCC组织中GSH水平降低(图S14C), MDA水平升高(图S14D)。
为了揭示更多的临床相关性,我们转向patient-derived异种移植(pdx)模型,已应用于临床前药物检测在许多类型的癌症由于其生物稳定,准确反映病人肿瘤组织病理学、基因表达、基因突变,和治疗反应(4G).