






YTHDF1通过以m6A依赖的方式促进肝细胞癌的进展
肝细胞癌(HCC)是最具侵袭性的恶性肿瘤之一,是全球第四大癌症相关死亡原因。新的证据表明,m6A在肿瘤进展中起着关键作用。然而,YTHDF1在HCC的生物学功能仍不清楚。发表于影响因子7.032的“Mol Ther Nucleic Acids”的“YTHDF1 Facilitates the Progression of Hepatocellular Carcinoma by Promoting FZD5 mRNA Translation in an m6A-Dependent Manner”详细介绍了YTHDF1在HCC中的生物学功能及作用机制。在这里,我们发现在HCC组织和细胞系中YTHDF1的表达显著升高,并且与HCC患者的预后显著相关。此外,在HCC中,YTHDF1的表达受USF1和c-MYC转录调控。功能研究表明,YTHDF1在体内外均可促进HCC细胞的增殖和转移。多组学分析显示,YTHDF1可以以m6A依赖的方式加速FZD5 mRNA的翻译输出,并通过WNT/β-catenin途径作为癌基因发挥作用。

技术路线:
结果:
(1) YTHDF1表达在HCC中显著上调并与不良预后相关
为了探索YTHDF1在HCC的调节作用,我们首先分析了它的表达。TCGA数据库、GSE14520数据集和GSE94660数据集显示YTHDF1在HCC组织中的表达显著上调(图1A)。此外,我们通过临床样本的qRT-PCR和免疫组织化学(IHC)分析检测了YTHDF1的表达,与相邻正常组织相比,HCC组织中YTHDF1在mRNA和蛋白水平上均过表达(图1B和1C)。此外,qRT-PCR分析证实,与LO2细胞相比,HCC细胞中的YTHDF1表达升高(图1D)。为探讨YTHDF1表达与临床病理特征的相关性,我们根据YTHDF1表达的中值将HCC患者分为两组。用Kaplan-Meier法进行的生存分析表明,高YTHDF1表达的HCC患者经历了更糟糕的OS(图1E)。有趣的是,与相应的正常组织相比,在多种实体恶性肿瘤中发现YTHDF1的表达显著上调(图1F)。根据上述数据,我们得出结论,YTHDF1的表达在HCC显著上调,并与HCC患者的不良预后相关。

(2) YTHDF1的表达由USF1和c-MYC共同控制
鉴于转录因子在基因表达中的广泛调节作用,我们旨在研究在YTHDF1的表达是否受到特定转录因子的调节。UCSC基因组浏览器数据库显示YTHDF1启动子在多种细胞系中非常活跃,包括HepG2细胞(图2A)。我们接下来分析了从ENCODE数据库下载的HepG2细胞ChIP-seq数据。如图2A所示,在YTHDF1启动子中c-MYC/MAX和USF1的潜在结合(图2A)。c-MYC/MAX和USF1位于E-box的290bp (图2B)。此外,荧光素酶活性显示,在HEK293T细胞中用c-MYC或USF1过表达载体共转染YTHDF1启动子报告子后,只有野生型推定结合位点增加了YTHDF1启动子的激活(图2C)。qRT-PCR和免疫印迹分析表明,c-MYC/USF1的缺失减少了但过表达增加了YTHDF1在基因和蛋白质水平上的表达,当c-MYC和USF1都受到调节时,这种表达更为有效(图2D和2E)。IHC分析显示,与ANTs相比,c-MYC和USF1在HCC组织中的表达显著上调,并与YTHDF1的表达呈正相关(图2F和2G)。综上所述,在HCC,YTHDF1的表达由USF1和c-MYC共同控制。

(3) YTHDF1促进HCC细胞体外增殖和转移
为了研究YTHDF1在HCC的生物学功能,我们设计了慢病毒介导的shYTHDF1和CRISPR-dCas9基因激活系统,分别在HCC细胞中敲除和过表达YTHDF1的表达(图3A和3B)。集落形成试验显示,YTHDF1敲除明显抑制,但YTHDF1过表达显著增强HCC细胞的集落形成能力(图3C)。CCK-8分析和EdU分析显示沉默YTHDF1可以抑制肝癌细胞的增殖,而上调YTHDF1可以促进肝癌细胞的增殖(图3D和3E)。Transwell显示,YTHDF1下调和上调分别显著抑制和提高了HCC细胞的迁移和侵入能力(图3F和3G)。上述结果表明YTHDF1在HCC细胞中发挥致癌作用。

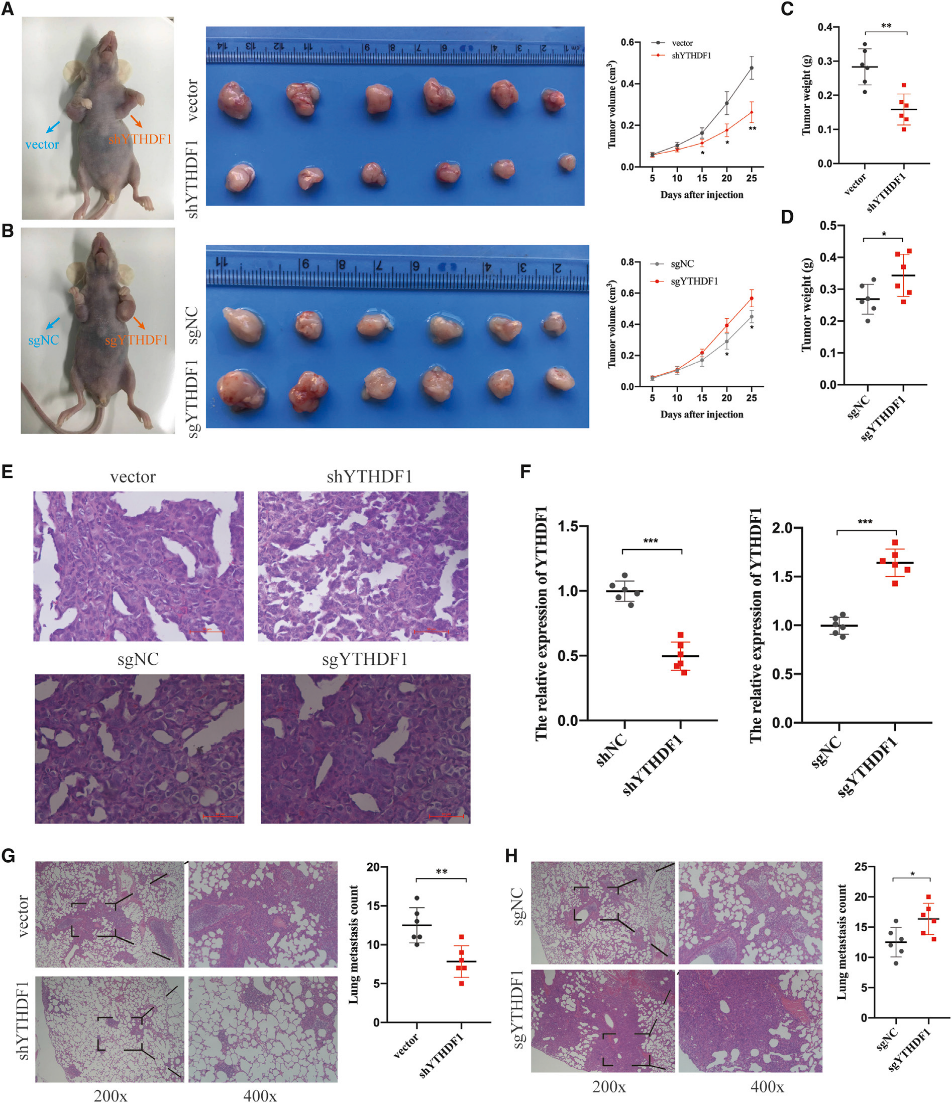
(4) YTHDF1促进体内HCC细胞增殖和转移
为了研究YTHDF1在HCC体内的致癌作用,我们用YTHDF1沉默的MHCC-LM3细胞和YTHDF1过表达的HepG2细胞在裸鼠体内进行了皮下植入实验。数据显示,YTHDF1敲除有效减少,但YTHDF1过表达显著增加肿瘤大小和重量(图4A-4D)。并通过HE染色验证各组肿瘤组织(图4E)。接下来,进行qPCR分析以确认异种移植肿瘤组织中YTHDF1的表达。正如预期的那样,YTHDF1沉默的MHCC-LM3细胞形成的肿瘤组织显示出降低的YTHDF1表达,而来自YTHDF1过表达的HepG2细胞的肿瘤组织显示出增加的YTHDF1表达(图4F)。为了评价YTHDF1是否能促进体内转移,我们通过静脉注射YTHDF1沉默的MHCC-LM3细胞和YTHDF1过表达的HepG2细胞建立了体内转移模型。我们观察到,与对照组相比,YTHDF1敲除组肺表面转移结节较少,而YTHDF1过表达组肺表面转移结节较多(图4G和4H)。总的来说,我们的发现揭示了YTHDF1促进体内HCC细胞的生长和转移。
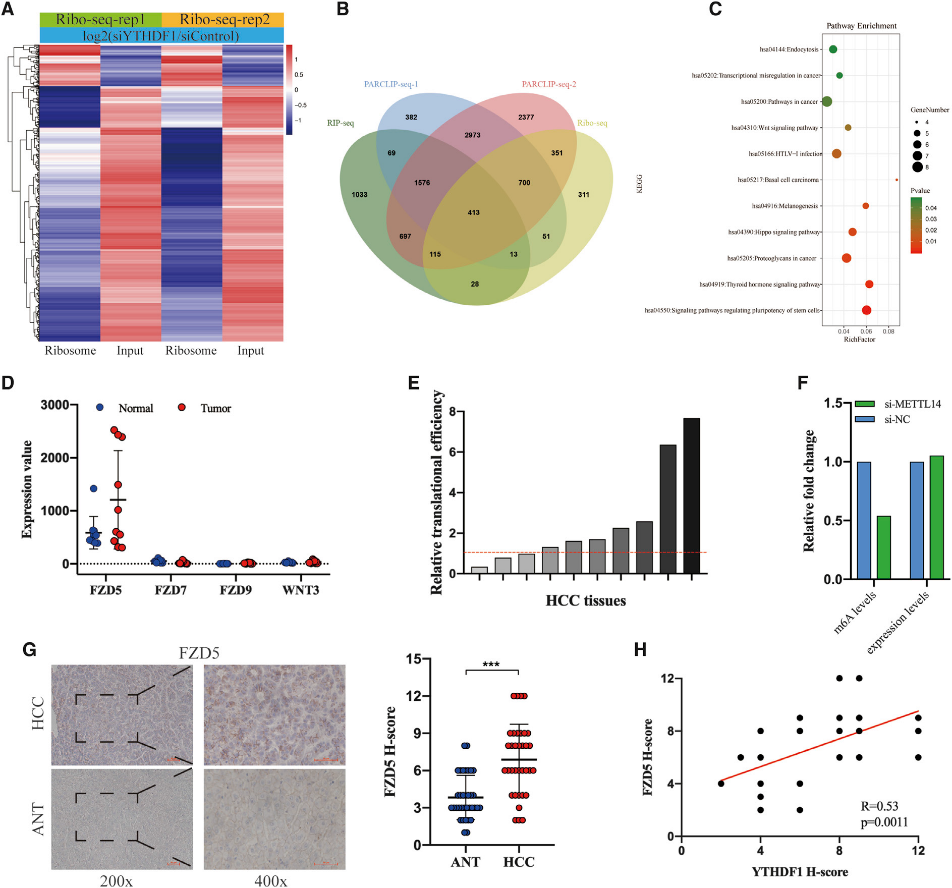
(5) 将FZD5确定为YTHDF1在HCC的直接目标
为了鉴定YTHDF1的靶标,我们首先分析了具有YTHDF1敲除的细胞的公共核糖体图谱数据(GSE 63591)。YTHDF1的缺失导致1982个转录物的翻译效率降低(图5A)。为了选择与YTHDF1直接结合的靶点,我们下载了GSE63591中YTHDF1的RNA RIP-seq和PARCLIP-seq数据,并与核糖体-seq数据重叠。我们发现YTHDF1的缺失导致与YTHDF1结合的413个基因的翻译效率降低(图5B)。KEGG分析表明,这些转录物在11种信号通路中显著富集,如Hippo和WNT信号通路(图5C),其中FZD5、FZD7、FZD9和WNT3在大多数通路中富集(8/11)。有趣的是,当我们使用GSE112705数据集在HCC中检测FZD5、FZD7、FZD9和WNT3的表达丰度和翻译效率时,我们发现FZD5的表达丰度比FZD7、FZD9和WNT3高得多,并且与ANTs相比,FZD5在大多数HCC组织中的转录效率得到提高(图5D和5E)。考虑到m6A的修饰具有高度的细胞类型特异性,我们分析了GSE9064212中的HepG2细胞RNA-seq和m6A-seq数据,以验证FZD5基因的m6A修饰,结果表明METTL14的敲除显著降低了人肝癌细胞中m6A的水平,而不是FZD5基因的表达水平(图5F)。IHC分析显示,与ANTs相比,FZD5在HCC组织中的表达显著上调(图5G),并且与YTHDF1的表达呈强正相关(图5H)。
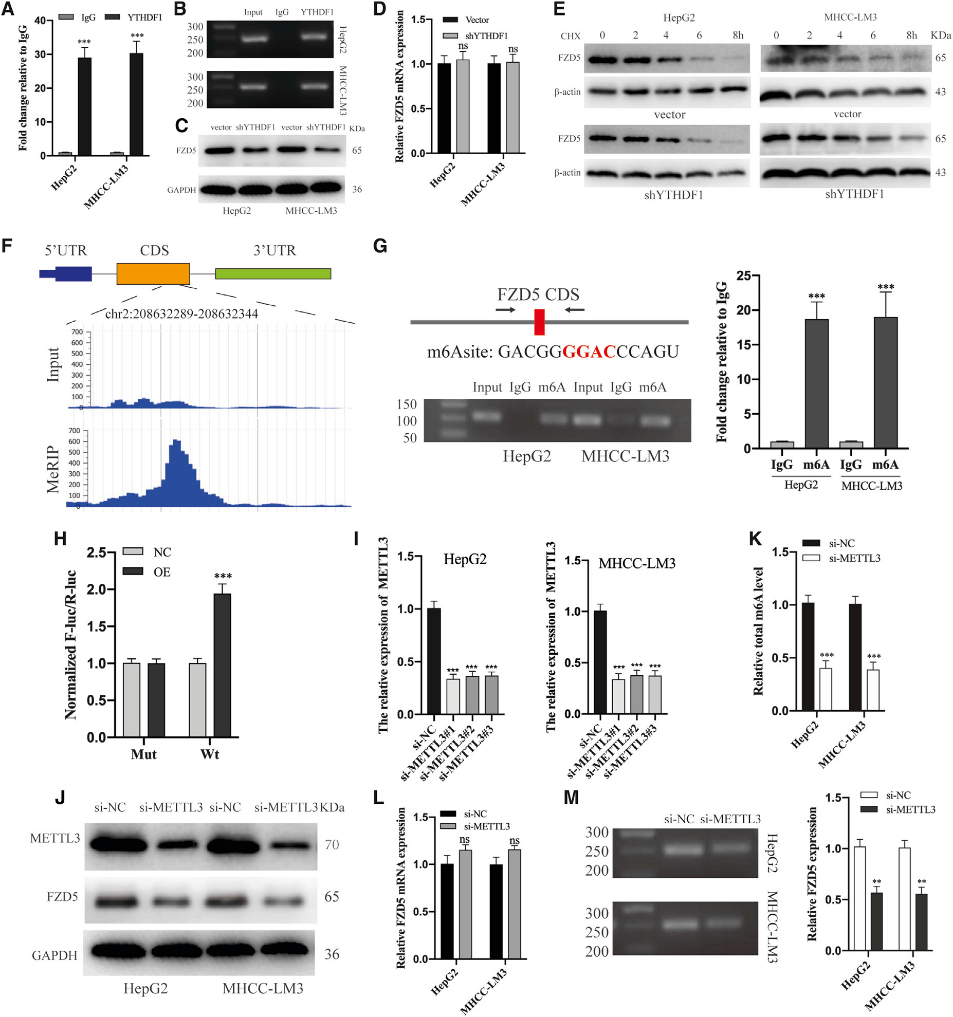
(6) YTHDF1以m6A依赖的方式促进FZD5 mRNA的翻译
为了验证FZD5基因和YTHDF1蛋白之间的直接结合,我们进行了RIP实验。半定量PCR和qRT-PCR分析显示与免疫球蛋白G相比,FZD5 mRNA与YTHDF1蛋白结合显著富集(图6A和6B)。此外,敲除YTHDF1导致FZD5在蛋白质水平上的显著抑制(图6C),但在mRNA水平上没有(图6D)。为了排除YTHDF1可能影响FZD5蛋白稳定性,我们用环己酰亚胺(CHX)处理HepG2和MHCC-LM3细胞以阻断翻译,并发现FZD5蛋白表达在载体组和shYTHDF1组中类似地被消除(图6E)。上述结果表明,由YTHDF1沉默引起的FZD5蛋白表达的消除是由于mRNA翻译效率的降低,而不是转录或蛋白稳定性的降低。
众所周知,YTHDF1通过结合m6A甲基化转录物发挥作用。因此,我们首先使用MeT-DB V2.0数据库筛选了HepG2细胞中FZD5基因的m6A位点,该数据库显示FZD5基因m6A位点富集在位于chr 2:208632313–208632314的蛋白质编码序列(CDS)中(图6F)。MeRIP-PCR证实了m6A修饰在该位点的显著富集(图6G)。此外,荧光素酶分析显示,YTHDF1过表达增强了FZD5-WT的表达(图6H)。此外,METTL3敲除(图6I和6J)显著降低了HCC细胞中m6A总水平(图6K)和FZD5蛋白表达(图6J),FZD5 mRNA表达略有上调(图6L)。此外,RIP分析显示,METTL3敲除显著抑制了YTHDF1和FZD5 mRNA之间的结合(图6M)。我们的结果表明,YTHDF1选择性地识别FZD5基因CDS中的m6A位点,并随后促进其翻译输出。
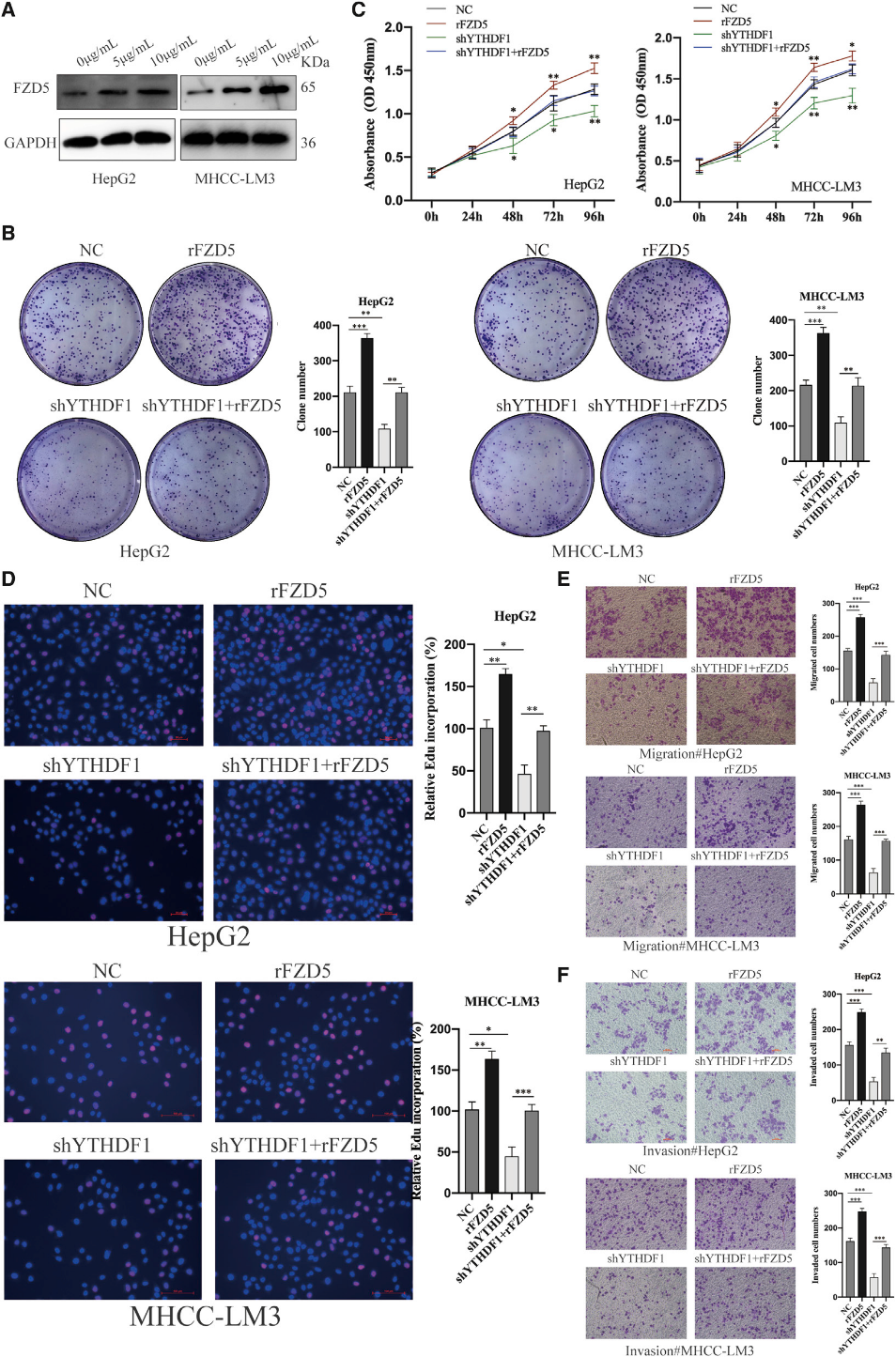
(7) FZD5过表达有效逆转YTHDF1敲除诱导的HCC细胞进程抑制
为了探索FZD5在HCC的调节作用,我们通过转染具有生物活性的重组人FZD5来过表达FZD5 (图7A)。细胞表型显示FZD5上调显著促进了HCC细胞的增殖、迁移和侵袭(图7B-7F)。接下来,我们在YTHDF1缺陷型HCC细胞中过表达FZD5,以研究YTHDF1的致癌作用是否由FZD5直接介导。集落形成试验、CCK-8试验和EdU试验表明,YTHDF1敲除损害了细胞集落形成和生长,而FZD5上调逆转了这种效应(图7B-7D)。一致地,Transwell分析显示,在HCC细胞中FZD5过表达后,由YTHDF1耗竭抑制的细胞迁移和侵入能力重新建立(图7E和7F)。
(8) YTHDF1通过WNT/β-Catenin信号通路促进HCC癌的发生
据报道,FZD5过表达在肝癌中激活WNT/β-Catenin途径。接下来,我们研究了YTHDF1是否通过FZD5/WNT/β-Catenin发挥作用。亚细胞分级和免疫印迹分析显示,YTHDF1的敲除显著降低了HCC细胞中β-Catenin的总表达和核表达,而FZD5的上调逆转了这种效应(图8A)。此外,我们发现YTHDF1敲除不影响细胞外钙的浓度(图8B)和磷酸化JNK水平(图8C)。这些结果证实了YTHDF1在HCC的致癌作用是通过WNT/β-Catenin途径而不是WNT/Ca2+途径或平面细胞极性途径介导的。众所周知,c-Myc基因是一种由WNT/β-Catenin信号转录激活的癌基因,它在很大程度上参与了各种癌症的发展。免疫组化分析表明,YTHDF消耗抑制但FZD5的过表达重建了HCC细胞中c-MYC的表达(图8A)。此外,IHC分析显示,与阴性对照组相比,FZD5、β-Catenin和c-MYC在YTHDF1下调的异种移植肿瘤样品中的表达显著降低(图8D)。此外,我们在HCC的TCGA数据库中观察到FZD5、β-Catenin、cMYC和YTHDF1表达呈正相关(图8E)。综上所述,我们的发现表明YTHDF1通过FZD5/WNT/β-Catenin信号通路促进HCC细胞的致癌作用(图8F)。

结论:YTHDF1可以通过FZD5/WNT/β-Catenin信号通路,以m6A依赖的方式提高FZD5 mRNA的翻译输出,促进HCC细胞的进程。我们的研究为HCC提供了一个潜在的治疗策略。