铁死亡损伤通过依赖TMEM173/STING途径促进胰腺肿瘤发生
胰腺癌约占美国所有癌症的3%和所有癌症死亡的7%。胰腺导管腺癌(PDAC)是一种外分泌肿瘤,是最常见的胰腺癌病理类型。尽管许多因素促使正常胰管向侵袭前病变转变为侵袭性PDAC,但这种进展主要是由炎症性肿瘤微环境中的内在Kras突变驱动的。了解胰腺肿瘤微环境的组成和功能可能有助于开发新的诊断和更好的治疗方法。
今天我们看的文章题名为:Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway,发表在nature communications杂志,影响因子12.121。改文章发现,高铁饮食或Gpx4消耗均可促进8-OHG释放,从而激活TMEM173 / STING依赖性DNA传感器途径,从而导致小鼠巨噬细胞浸润和激活Kras驱动的PDAC。
高铁饮食或Gpx4缺失促进实验性胰腺炎
胰腺炎是胰腺癌发生的危险因素,首先研究了铁死亡对实验性急性胰腺炎模型的影响,急性胰腺炎模型是由雨蛙肽或L-精氨酸诱导的。高铁饮食3个月后,小鼠胰铁含量高于对照组。与对照饮食小鼠相比,高铁饮食小鼠更易受雨蛙肽或L-精氨酸诱导的胰腺炎的影响,死亡率显著更高。HE评估显示高铁饮食组腺泡细胞过度死亡、白细胞浸润和间质水肿。高铁饮食组的血清淀粉酶、胰腺胰蛋白酶活性和胰腺髓过氧化物酶活性显著升高,表明高铁水平加速了胰腺炎的进展。
接下来确定GPX4介导的抗氧化反应对实验性胰腺炎的影响。小鼠胰腺中Gpx4的条件性KO不影响胰腺发育和内分泌功能和铁水平,但会导致雨蛙肽或L-精氨酸诱导的胰腺炎比野生型(WT)小鼠发展更快,死亡率、胰腺损伤和胰腺炎相关酶(淀粉酶、胰蛋白酶和髓过氧化物酶)增加。相反,服用liproxstatin-1(一种铁死亡抑制剂),对雨蛙肽或L-精氨酸诱导的胰腺炎有保护作用,特别是在高铁饮食或Gpx4缺乏的情况下。这提示铁过载和Gpx4缺失通过氧化损伤加速实验性胰腺炎的进展。

高铁饮食或Gpx4缺失促进Kras驱动的胰腺肿瘤发生
与KC(Pdx1-Cre; KrasG12D/+)小鼠相比,Gpx4缺失KCG(Pdx1-Cre;KrasG12D/+,Gpx4−/−)小鼠高铁饮食进一步增加Kras介导的动物死亡,胰腺重量增加,胰腺上皮内瘤变(PanIN)的形成和基质反应。相比之下,Gpx4耗竭或高铁饮食会减少正常的腺泡细胞。KC小鼠的胰腺铁高于对照饮食。正常饮食的KC和KCG小鼠之间的胰腺铁无差异。在10-12个月大时,在KCG小鼠中观察到肿瘤侵袭或向肝和肺转移的增加。这些发现表明高铁饮食或Gpx4缺失会加速胰腺Kras介导的PDAC。接下来研究了Gpx4缺失或高铁饮食对Kras驱动的分子事件的影响,这些分子事件涉及腺泡到导管的化生、导管病变、基质反应和转移。腺泡到导管化生(Sox9)、导管病变(Krt19)、基质反应(Vim)和转移(Mmp9)通过Gpx4缺失或高铁饮食上调。
相反,铁死亡抑制剂liproxstatin-1对Kras引起的动物死亡以及Gpx4缺失或高铁饮食小鼠的病理和分子变化具有保护作用。此外,在Kras引起的胰腺肿瘤发生中,Gpx4缺失或高铁饮食均上调了胰腺中PTGS2(一种铁下垂的标志物)和Ki67(一种肿瘤细胞增殖的标志物)的表达。这些动物研究表明,铁死亡可能促进Kras驱动的胰腺肿瘤的发生。值得注意的是,杂合子缺失Gpx4未能加速Kras介导的动物死亡、胰腺导管上皮内瘤变和基质反应。在3个月大时,纯合Gpx4缺失和高铁饮食对Kras介导的胰腺导管癌变和基质反应有协同作用。

巨噬细胞耗竭减少胰腺肿瘤发生
鉴于巨噬细胞是胰腺肿瘤发生早期的主要参与者,接下来测试了Gpx4缺失或高铁饮食对肿瘤微环境中巨噬细胞浸润和激活的影响。F4/80免疫荧光染色观察巨噬细胞浸润情况。肿瘤相关巨噬细胞(TAM)在Kras驱动的小鼠中随着Gpx4缺失或高铁饮食而增加,表明铁和Gpx4是胰腺肿瘤微环境中巨噬细胞浸润的调节因子。为了进一步确定巨噬细胞在高铁饮食或Gpx4消耗依赖性肿瘤发生中的作用,使用氯膦酸盐脂质体来消耗巨噬细胞。与对照脂质体相比,氯膦酸盐脂质体对巨噬细胞抑制作用降低了Gpx4缺失(KCG)或高铁饮食小鼠的动物死亡率、胰腺重量、癌变和基质反应;Sox9、Krt19、Vim和Mmp9的表达被氯膦酸盐脂质体下调。因此,巨噬细胞聚集似乎是高铁饮食或Gpx4缺失依赖性胰腺肿瘤发生所必需的。

氧化核碱基通过TMEM173诱导巨噬细胞迁移和活化
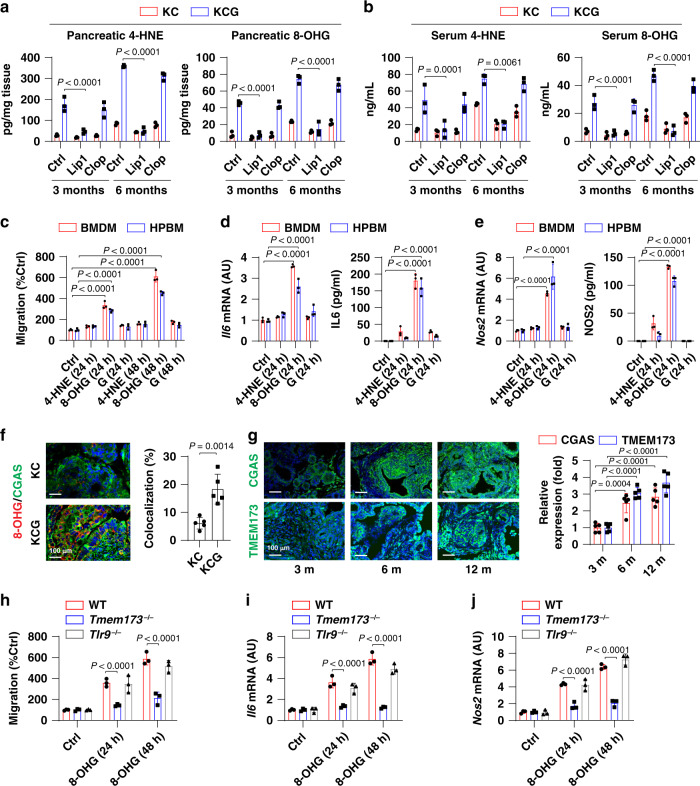
Gpx4耗尽或高铁饮食导致氧化胰腺或血清中DAMP的产生和释放增加,DAMP包括4-HNE(脂质过氧化产物)和8-OHG(DNA氧化损伤的主要产物)。接下来测试了这些氧化损伤相关的DAMP是否能在体外调节巨噬细胞的迁移和活化。与4-HNE相比,8-OHG(500 ng/ml)显著诱导原代小鼠骨髓源性巨噬细胞(BMDM)或人血单核细胞源性巨噬细胞(HPBMs)中的细胞迁移和细胞因子Il6和iNos表达释放。这些结果表明氧化的碱基(8-OHG)而不是氧化的脂质(4-HNE)可能促进巨噬细胞的迁移和活化。
鉴于8-OHG是一种氧化的DNA损伤产物,然后测试了TMEM173是否是宿主细胞中的一种主要的DNA感应调节剂,调节8-OHG活性。DNA传感器环GMP-AMP合酶(CGAS)与宿主DNA结合,启动TMEM173依赖性反应。与对照组KC小鼠相比,来自KCG小鼠的肿瘤中8-OHG DNA和CGAS的共定位增加,支持先前的发现8-OHG是CGAS的直接配体。在Kras驱动的小鼠PDAC期间,肿瘤微环境中TMEM173或CGAS的表达以时间依赖性方式增加。在体外,Tmem173的缺失阻断了8-OHG诱导的细胞迁移和BMDM中Il6和Nos2的mRNA表达。这些数据表明8-OHG诱导的巨噬细胞迁移和活化需要TMEM173,而不是TLR9。
TMEM173促进胰腺肿瘤发生
为了确定TMEM173通路是否是胰腺癌发生所必需的,首先将8-OHG抗体注射到KGC小鼠体内。与IgG对照相比, 8-OHG抗体延长了动物存活时间,抑制了Gpx4缺失介导的胰腺增重和快速肿瘤进展,减少了肿瘤形成,减少了基质反应,减少了TAM浸润。
在KC小鼠中,Tmem173的缺失可防止Gpx4缺失诱导的动物死亡、肿瘤进展和TAM浸润减少。阻断8-OHG-TMEM173通路也减少了Gpx4缺失诱导的胰腺中Sox9、Krt19、Vim和Mmp9的上调。此外,给予8-OHG抗体或去除Tmem173可防止高铁饮食诱导的动物死亡和肿瘤进展,以及Kras驱动小鼠中的TAM浸润。8-OHG的过度积累可能促进染色体的不稳定性,如端粒异常。与对照KCG小鼠相比,端粒FISH分析表明,KCGT小鼠中Tmem173的缺失并未显著改变端粒缺失。有趣的是,在KCG和高铁饮食小鼠中,通过氯膦酸盐脂质体消耗巨噬细胞减少TMEM173表达,这表明巨噬细胞浸润、TMEM173激活和铁死亡之间存在反馈机制。这些动物研究表明8-OHG-TMEM173通路的激活以端粒损伤独立的方式促进胰腺肿瘤的发生。

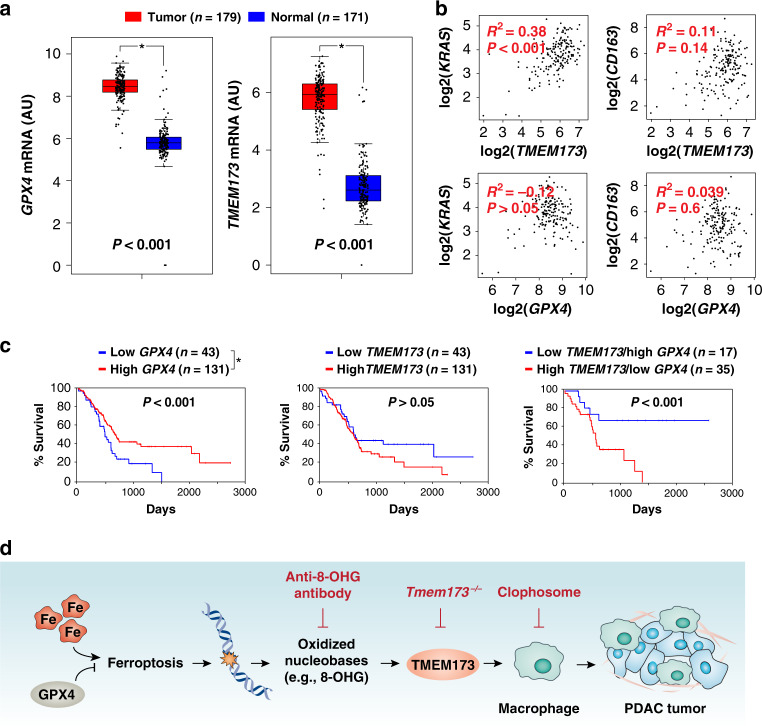
GPX4是人PDAC的预后标志物
动物研究表明GPX4可能在胰腺癌中起到抑癌作用,而TMEM173可能具有致癌作用。使用TCGA数据库分析,与正常组相比,PDAC组的GPX4和TMEM173 mRNA表达均上调。在PDAC患者中,KRAS和TMEM173的mRNA表达也呈正相关,KRAS和GPX4的mRNA表达之间没有显著相关性。尽管巨噬细胞耗竭降低了小鼠肿瘤微环境中TMEM173的表达,但在PDAC患者中GPX4和TMEM173的mRNA表达与巨噬细胞标记物CD163之间没有显著相关性。总生存率分析进一步显示,GPX4的高表达与PDAC患者的生存率增加相关。TMEM173的mRNA表达与PDAC患者的总生存率之间没有显著相关性。此外,GPX4的低表达和TMEM173的高表达增加了PDAC患者的死亡率。这些分析表明GPX4可能是人类PDAC的主要预后指标。