






CDK4/6抑制通过诱导T细胞记忆促进抗肿瘤免疫
细胞周期蛋白依赖性激酶4和6(CDK4/6)的药理学抑制剂是激素受体阳性乳腺癌的获批治疗药物,目前正在其他癌症类型的数百项临床试验中进行评价。这些抑制剂的临床成功在很大程度上归因于定义明确的肿瘤内在细胞抑制机制,而其作为免疫调节剂的新作用知之甚少。本研究利用整合的表观基因组、转录组学和蛋白质组学分析,证明了CDK4/6抑制剂在促进免疫T细胞记忆的表型和功能获得中的新作用。本文的机制见解拓宽了CDK4/6抑制剂作为增强抗肿瘤T细胞免疫的临床工具的前瞻性效用。本研究于2021年5月14日发表在《Cancer Discovery》IF:29.497期刊上。
技术路线:

主要实验结果:
1、在响应CDK4/6抑制中瘤内记忆类CD8+ T细胞的表现
为了全面评价CDK4/6抑制对抗肿瘤T细胞免疫的影响,使用多模式单细胞测序(CITE-seq)分析了CDK4/6抑制剂palbociclib或对照处理的MC38-OVA肿瘤小鼠的瘤内T细胞群体 (Fig. 1A)。维度分析显示MC38-OVA肿瘤中有10个不同的T细胞群(Fig. 1B)。用CDK4/6i处理的肿瘤有较低频率的CD4+调节性T细胞(Foxp3+ Tregs),而CD8+记忆类T细胞频率更高,以Tcf7、Sell、Bcl2和Cd7高表达为特征(Fig. 1B-D)。CD8+ T细胞池的基因动态表达揭示来源于CDK4/6i处理的小鼠的CD8+肿瘤免疫浸润(TILs)向更早、更像干细胞的分化状态倾斜(Fig. 1E-F)。与此一致,较早出现假时间轨迹的细胞表达更高水平的记忆相关基因(Tcf7, Sell, Il7r),和更低水平的效应相关基因(Prf1, Gzmb)和耗损标志物(PD-1, TIM3) (Fig. 1G)。支持了转录组学分析,流式细胞术表明带有中央记忆(Tcm)表型(CD44+CD62L+)的CD8+ TILs的比例显著更高,这在不同的肿瘤模型中是一致的(Fig. 1H)。为了检验抑制CDK4/6对抗肿瘤T细胞免疫的长期功能效应,用CDK4/6i在短时间 (抗肿瘤T细胞反应最活跃的3-13天)处理MC38-OVA荷瘤小鼠,然后停药。在停止治疗时,CDK4/6i处理小鼠和对照小鼠的肿瘤体积是相等的(Fig. 1I-J)。引人注目的是,在停止治疗后,既往使用CDK4/6i治疗的小鼠中有21只小鼠肿瘤完全清除,而对照组只有9只(Fig. 1K)。总之,这些数据表明CDK4/6的药理抑制促进T细胞记忆,并产生能够驱动有效和持续的抗肿瘤反应的瘤内T细胞室。

图1用CDK4/6抑制剂处理的小鼠出现瘤内记忆类CD8+ T的细胞。
2、CDK4/6i介导的CD8+ T细胞记忆获得是细胞固有的
为了确定在CDK4/6i处理的肿瘤中观察到的T细胞记忆是否由于CDK4/6在这些细胞内的直接抑制,在体外将活化的小鼠CD8+ T细胞暴露于CDK4/6i中,并监测Tcm获取和分裂数。激活CDK4/6i暴露0、24、72 h后,Tcm细胞平均分裂数减少,同时细胞比例增加,且呈剂量依赖性,仅在24 h内发生(Fig. 2A)。这表明CDK4/6i不是简单地抑制分化,而是直接促进记忆形成,表明细胞周期机制与分化之间存在方向性关系。通过3'RNA-seq进一步分析发现,在CDK4/6i处理后,记忆相关转录本增加,GSEA分析证实了记忆相关特征的富集,以及细胞周期相关特征的减少,包括E2F靶点(Fig. 2B-E)。矛盾的是,CDK4/6抑制也诱导了效应相关转录本,包括Gzma、Gzmc和Pdcd1 (Fig. 2B-C),这与之前CDK4/6i促进T细胞活化的报道一致。
基序分析发现E2F TF基序在乙酰化降低相关区域显著富集,在T细胞记忆驱动因子相关基序乙酰化增加,包括含叉头框因子(Fig. 2F)。为了确定这些变化是否伴随着对不同细胞状态的长期表观遗传影响,在CDK4/6抑制后对体外活化的T细胞进行了ATAC-seq,观察到染色质开放性的整体降低,T细胞记忆相关基因区域的开放性增加,包括Tcf7、Ccr7和Sell (Fig. 2G)。为了解开CDK4/6抑制后记忆和效应特征的明显二分法,在暴露于CDK4/6i 24h后,对体外活化T细胞进行了单细胞RNA-seq。该分析显示存在CDK4/6i处理后改变的不同T细胞亚群,簇2、4、5和8增加,簇0减少,以及整体G1细胞周期停滞(Fig. 2H-I)。接下来对记忆和效应基因标记,并使用AUCell比较富集评分,确定细胞池中不同的记忆样和效应样簇(Fig. 2J)。有趣的是,CDK4/6i处理后增加的细胞簇2和5是记忆样细胞,8是效应样细胞,证明CDK4/6i抑制促进异质T细胞池中表型不同的亚群细胞的记忆分化,增强效应功能。

图2 CDK4/6i介导的CD8+ T细胞记忆获得是细胞固有的
3、CDK4/6抑制剂促进记忆形成通过RB介导的G1停滞
为了确定驱动CDK4/6i诱导的记忆,对调节记忆标记的基因CD62L (SELL),进行了全基因组CRISPR/Cas9筛选,用基因组范围的sgRNA文库转导表达Cas9的Jurkat T细胞(CDK4/6抑制后上调CD62L),然后用CDK4/6i处理,并对未能上调CD62L的细胞进行荧光激活细胞分选(Fig. 3A)。对分选群体进行测序发现,靶向RB转录共抑制因子1(RB1)的sgRNA仅在处理条件下显著富集(Fig. 3B),暗示RB是CDK4/6i诱导CD62L上调所必须的。随后对急性(2h)暴露于CDK4/6i后的Jurkat细胞和活化的原代小鼠CD8 + T细胞进行了整体磷酸化蛋白质组学分析(Fig. 3C)。在Jurkat和小鼠T细胞中检测到多个肽上特异性磷酸化位点的显著缺失,包括典型的CDK4/6靶标RB和RBL 1/2 (Fig. 3D-F)。值得注意的是,RB在两种细胞蛋白组学中重叠,并且是全基因组CRISPR/Cas9筛选最显著的(Fig. 3B,3G),表明CDK4/6i介导的记忆形成是由RB介导。因此,靶向敲除RB1,可以消除CD62L的表达上调(Fig. 3H)。3’RNA-seq分析显示RB缺失废除了转录对CDK4/6i的响应,包括SELL,其被CDK4/6i诱导的转录改变被RB缺失所大幅度减弱了(Fig. 3I)。与此一致,靶向敲除RB1也废除了激活的初级小鼠CD8+T细胞的Tcm形成(Fig. 3J)。
为了确定CDK4 /6i诱导的记忆形成是否通过细胞周期的RB介导的G1期阻滞,用G1阻滞剂处理细胞,胸腺嘧啶,通过阻止DNA合成和进入S期,独立于RB的诱导G1阻滞。结果显示胸腺嘧啶处理的细胞表型复制了CDK4/6抑制,并在很大程度上挽救了Rb1 KO细胞中Tcm的形成(Fig. 3J) ,表明RB介导的G1阻滞本身有助于CDK4 /6i诱导的记忆分化。通过3'RNA-seq进一步分析Rb1 KO细胞,发现记忆相关基因下调和效应标记增加(图3K-L),CDK4/6抑制后未能逆转(图3M-N)。这些结果表明CDK4/6i介导的转录重编程和记忆形成是通过RB依赖的G1细胞周期停滞和同时抑制CDK4/6信号转导。

图3 CDK4/6抑制剂促进记忆形成通过RB
4、CDK4/6预处理增强功能性记忆CD8+ T细胞的持久性
长期生存能力是T细胞记忆的基本特征。为了确定CDK4/6i处理的细胞在体内是否表现出优越的存活率,将未处理和CDK4/6i预处理的体外活化CD45.1 OT-i T细胞转移至同源CD45.2小鼠中,并通过流式细胞术评价其随时间的持续性和表型(Fig. 4A)。在30天内,在血液和脾脏中检测到的预处理OT-I - T细胞的频率明显更高(Fig. 4B)。30天后,未处理和预处理的OT-I - T细胞在血液和脾脏中的表型持久性相似,都具有记忆前体的特征 (MPECs; KLRG1-CD127+) (Fig. 4C)。为了评价CDK4/6i处理细胞的记忆能力,将未处理或体外活化的CD45.1 OT-i T细胞中预处理的细胞再次转移至同源CD45.2宿主中。30d后,分离出OT-ⅠT细胞,等量重新转移到同时感染李斯特菌-OVA(LM-OVA)的新宿主中(Fig. 4D)。未处理和预处理的OT-I T细胞在LM-OVA感染后扩增和分化,迅速获得功能性、产生细胞因子的SLEC表型(KLRG1 + CD127-)(Fig. 4E-F)。这些表明CDK4/6抑制驱动T细胞记忆的表型和功能获得。
然后使用单细胞RNA-seq来描述移植后30天脾脏中存留的T细胞的转录谱(Fig. 4G)。使用pseudobulk RNA-seq分析和预转移体外培养的基因比较,生成了持续细胞的基因标记,其标志是记忆相关基因(Sell,Foxp1,Tcf7,Cd7,Il7r,Klf2,Ccl5)的高表达和效应相关基因(Gzmd,Ifng,Prf1,Gzma,Gzmb)的低表达(Fig. 4H-I)。对先前的体外单细胞数据(Fig. 2H-J)的进一步分析显示,CDK4/6i处理后,具有这种持续性特征的细胞频率从7.5%增加至26.8%(Fig. 4J-K)。这些数据表明,抑制CDK4/6促进T细胞向真实的记忆表型分化,其特征是功能的长期持久性。
图4CDK4/6预处理增强功能性记忆CD8+ T细胞的持久性
5、CDK4/6预处理增强CAR-T细胞的持久性和有效性
接下来测试CDK4/6i处理是否会诱导嵌合抗原受体(CAR)-T细胞的记忆表型,并克服CAR-T细胞治疗成功的两大障碍:T细胞耗竭和缺乏持久性。通过临床试验,以Lewis Y (LeY)抗原为靶点,制造了人类CAR-T细胞 (Fig. 5A)并发现暴露于CDK4/6i产生CD4+和CD8+干细胞记忆(Tscm)CAR-T细胞 (Fig. 5B)。CAR-T细胞的3’RNA-seq揭示CDK4/6抑制后记忆表达显著改变,GSEA分析证实了记忆相对效应信号的富集,并如预期E2F靶基因下调 (Fig. 5C)。为了检测在体内的持久性,使用两个独立供体的PBMC生成的LeY CAR-T细胞用CDK4/6i预处理,并转移到NSG小鼠中(Fig. 5D)。与未经处理的对照组相比,观察到在移植后的几周内,血液中CD8+和CD4+预处理的CAR-T细胞的频率和数量显著增加(Fig. 5E-F)。为了评估这些CAR-T细胞的功能性记忆能力,在CAR-T细胞转移后30天用LeY + 卵巢癌细胞系OVCAR-3(Fig. 5D)处理小鼠,观察到与未处理的CAR组相比,预处理的CAR-T细胞在小鼠血液中的扩增显著增加(Fig. 5G)。意味着先前接受过CAR-T细胞治疗的小鼠的肿瘤控制显著增强(Fig. 5H-I)。值得注意的是,第30天的肿瘤大小与肿瘤侵袭前血液中CD3+ T细胞的数量呈显著负相关(Fig. 5J),表明CDK4/6i处理的CAR-T细胞的持久性增强是驱动肿瘤控制的关键因素。事实上,肿瘤接种后40天,接受预处理的CAR-T细胞的小鼠浸润CD4+和CD8+ CAR-T细胞的数量显著增加(Fig. 5K)。接下来通过将未处理或预处理的抗LeY CAR-T细胞转移到OVCAR-3荷瘤小鼠体内,检测了CDK4/6i预处理对CAR-T细胞急性疗效的影响。用CDK4/6i预处理CAR-T细胞可显著增强其抗肿瘤活性(Fig.5L-M)。与此一致,转移后数周发现接受预处理细胞的小鼠血液中CD4 + 和CD8 + 细胞以及肿瘤中CD8 + 细胞数量显著增高(Fig.5N-O)。总之,这些数据证明CDK4/6i体外治疗是增强CAR-T细胞表型和长期疗效的稳健策略。

图5 CDK4/6预处理增强CAR-T细胞的持久性和有效性
6、CDK4/6抑制诱导T细胞表型与免疫检查点封锁的良好反应相关
在临床小鼠模型中,CDK4/6抑制剂与ICB显示出显著的协同作用。鉴于最近的研究强调肿瘤微环境中的干细胞样T细胞是ICB反应的关键介质,作者假设CDK4/6i介导的T细胞记忆诱导产生了更有利的T细胞池用于免疫检查点靶向治疗,因此将有助于该联合治疗的疗效。事实上,这种CDK4/6i应答特征富集在ICB应答患者的CD8 + TIL中,在单细胞和患者水平准确分为应答者和非应答者(Fig.6A-D)。这表明CDK4/6i诱导了T细胞内基因特征,该特征与患者对ICB的良好反应相关。
作者收集并分析了黑色素瘤患者在治疗过程中的连续血液样本,在此期间,患者接受CDK4/6i联合靶向MEK抑制剂治疗,随后接受PD-1和CTLA-4 ICB联合治疗(Fig.6 F),患者达到完全缓解。使用CITE-seq技术评估一系列患者样本,观察到CDK4/6i + MEKi治疗后CD8 + T细胞记忆群的频率呈时间依赖性增加,其特征为SELL、IL7R和TCF7的高表达(Fig.6G-I),这与临床分析一致。事实上,伪时间分析揭示了从记忆样群体到效应细胞的分化轨迹,CDK4/6i在该轨迹早期抑制CD8 + T细胞,随后的ICB治疗加速了分化(Fig.6J)。跨越该时间的TCR克隆追踪也发现,CDK4/6抑制增加了罕见T细胞克隆的频率,主要存在于记忆群体内,随后ICB处理扩增了这些克隆(Fig.6K)。这表明ICB治疗释放了外周血中更多种类T细胞受体。总之,这些数据表明,CDK4/6i可用作ICB给药前促进更有利T细胞表型的启动工具。

这项研究证明了在细胞毒性T细胞中药理学抑制CDK4/6可诱导RB介导的G1期阻滞,并促进记忆表型的获得,可显著增强这些细胞的长期抗肿瘤活性(Fig. 7)。
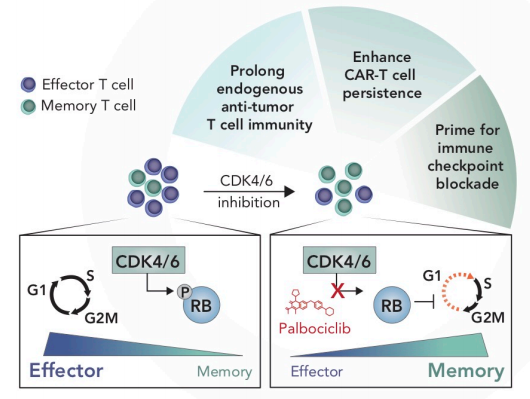
图7 CD8+ T细胞CDK4/6抑制剂的免疫增强效应模型
参考文献:
Lelliott Emily J., Kong Isabella Y., Zethoven Magnus., Ramsbottom Kelly M., Martelotto Luciano G., Meyran Deborah., Jiang Zhu Joe., Costacurta Matteo., Kirby Laura., Sandow Jarrod J., Lim Lydia., Dominguez Pilar M., Todorovski Izabela., Haynes Nicole M., Beavis Paul A., Neeson Paul J., Hawkins Edwin D., McArthur Grant A., Parish Ian A., Johnstone Ricky W., Oliaro Jane., Sheppard Karen E., Kearney Conor J., Vervoort Stephin J.(2021). CDK4/6 inhibition promotes anti-tumor immunity through the induction of T cell memory. Cancer Discov, undefined(undefined), undefined. doi:10.1158/2159-8290.CD-20-1554