






Rank-In:芯片、转录组数据综合分析平台

尽管转录组学技术在过去几十年中发展迅速,但由于芯片和 RNA-seq 之间固有的可变性差异,对混合数据的综合分析仍然具有挑战性。本文提出了 Rank-In (http://www.baddcao.net/rank-in/index.html)来纠正两种技术之间的非生物效应,从而能够自由混合数据以进行整合分析。
网站首页如下,支持上传用户自己的芯片、转录组数据。该网页最终会给出调整后的矩阵、差异基因列表以及聚类结果图。

第一步:上传表达文件
表达文件格式如下,需上传第一行为样本名、第一列为基因名的表达矩阵
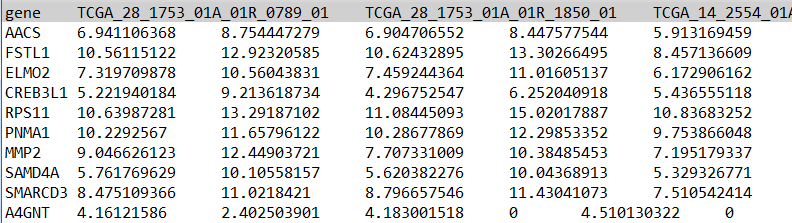
转录组数据表达量可以使用FPKM、TPM或TMM;芯片数据使用基因表达强度;如果下载GEO数据,需要对探针注释为基因,然后上传注释后的表达文件。
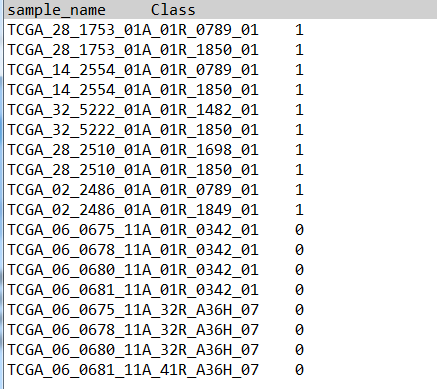
第二步:上传分组文件

分组文件第一列文样本名,第二列为分组。“1”表示来自正常组织的样本,“2”表示癌症亚型1的样本,“3”表示癌症亚型2的样本,依此类推
第三步:上传附加文件(可以选择性上传)

附加文件中可以包括平台、批次等信息。 例如,在平台列中,“1”表示数据来自 Affymetrix U133 plus2 平台,“2”表示来自安捷伦微阵列芯片的数据,“3”表示来自 Illumina Hiseq 2000 的数据等。
第四步:填写电子邮箱(选填,建议填写)
如果填写邮箱,结果会以邮件形式发送至该邮箱。
第五步:提交

数据文件全部上传后,点击“Submit”进行提交,页面会自动跳转至结果页。也可以根据页面给出的job ID查询结果。

结果页面如下:
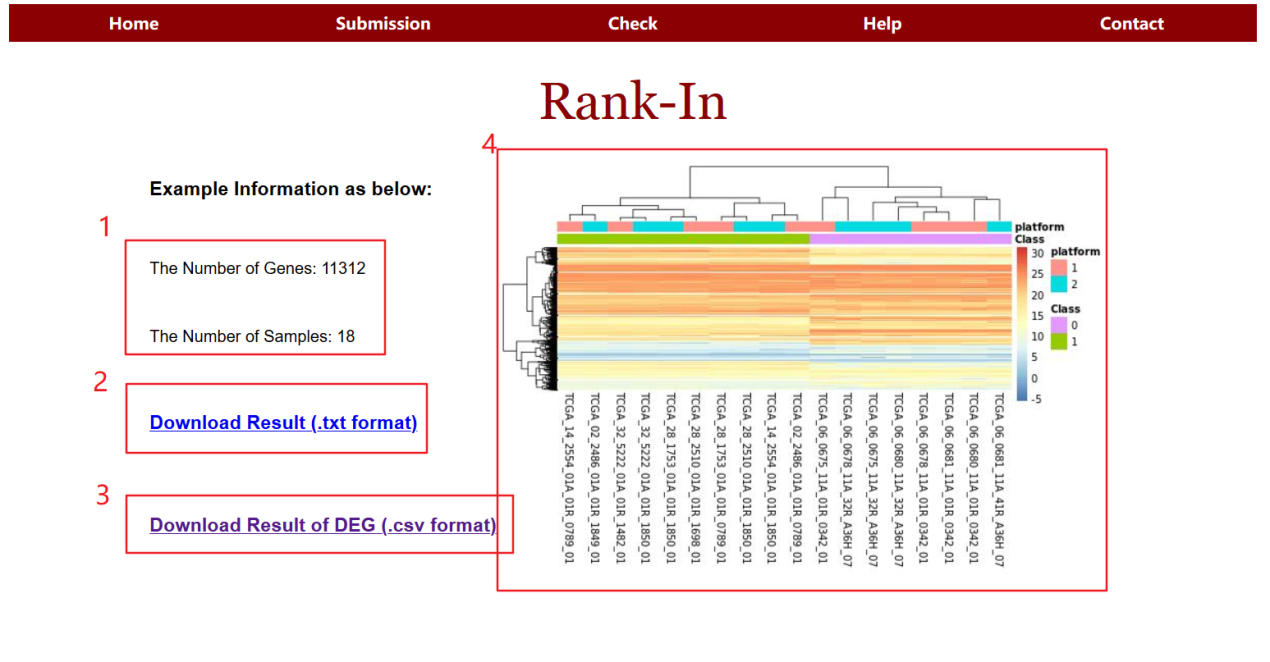
总而言之,我们可以使用Rank-In对癌症的芯片和 RNA-seq中的混合数据进行综合转录组学分析。