






中性粒细胞外泌体miR-30d-5p在脓毒症相关急性肺损伤中诱导M1巨噬细胞极化并诱导巨噬细胞焦亡

多形核中性粒细胞(PMN)在脓毒症相关急性肺损伤(ALI)中起重要作用。越来越多的证据表明,PMN衍生的外泌体是一种新的亚细胞实体,在PMN引发的炎症和组织损伤之间起着基础性的联系。然而,PMN衍生的外泌体在脓毒症相关ALI中的作用及其潜在机制尚不清楚。目前有研究表明,来自中性粒细胞的外体miR-30d-5p通过激活NF-κB诱导M1巨噬细胞极化和引发巨噬细胞焦亡而参与脓毒症相关的ALI。该研究于2021年10月发表在《Crit Care》,IF:9.097。
技术路线:
主要研究结果:
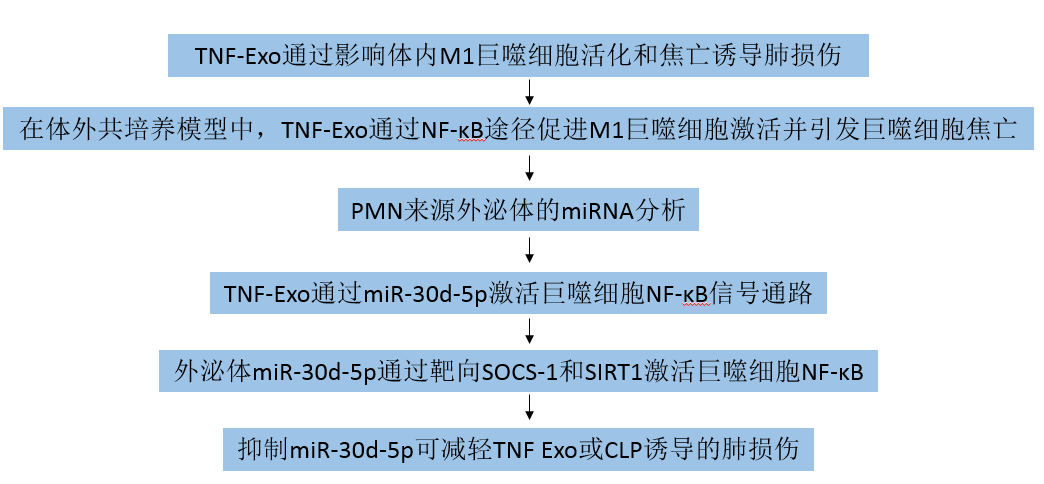
1. TNF-Exo通过影响体内M1巨噬细胞活化和焦亡诱导肺损伤
TNF-α是一种有效的炎症反应诱导因子,也是与败血症相关ALI中先天免疫的关键调节因子,常用于激活中性粒细胞。因此,用PBS(PBS-Exo)处理或用TNF-α(TNF-Exo)体外刺激C57BL/6J小鼠的中性粒细胞用来分离外泌体。荧光成像显示,注射TNF-Exo后,TNF-Exo在肺组织中积聚(图1a)。与PBS-Exo相比,TNF-Exo给药后小鼠肺部观察到组织学病变(图1b)。同时,肺组织中的促炎介质(iNOS、IL-1β和TNF-α)高度表达,局部M1巨噬细胞(iNOS+F4/80+细胞)数量显著增加(图1c,d),表明TNF-Exo诱导的肺炎症与巨噬细胞炎症活动一致。此外,研究发现TNF-Exo显著增加了腹腔巨噬细胞(PMφ)中M1巨噬细胞(CD11c+CD206-)和死亡细胞(膜联蛋白V+PI+)的比例(图1e,f)。为了进一步确定PMφ死亡的类型,通过流式细胞术检测PMφ的核碎裂、caspase-1激活(焦亡的特征),结果显示TNF-Exo注射后24小时PMφ的焦亡率约为8%(图1g)。所有这些数据表明,所有这些数据表明,TNF-Exo在体内可能由于M1巨噬细胞的活化和焦亡而诱发肺部炎症。
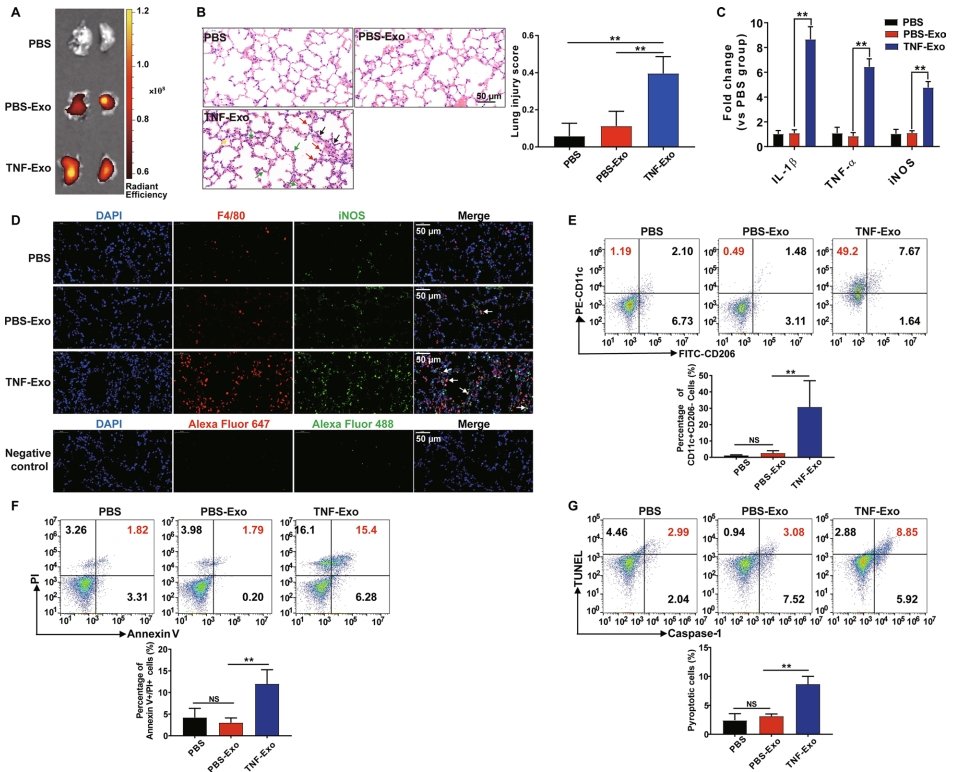
图1 TNF-Exo在体内通过影响M1巨噬细胞的活化和焦亡诱导肺损伤
2. 在体外共培养模型中,TNF-Exo通过NF-κB途径促进M1巨噬细胞激活并引发巨噬细胞焦亡
使用体外共培养系统,将巨噬细胞与PMN衍生的外泌体共培养,以进一步证实体内动物研究的结果。结果发现,无论是在原代细胞还是巨噬细胞细胞系中,TNF-Exo都能促进巨噬细胞M1极化(图2a-c)。然而,TNF-Exo并不能直接促进巨噬细胞死亡或焦亡;添加ATP/nigericin后,TNF-Exo诱导的巨噬细胞的焦亡性细胞死亡显著上调(图2d-e)。激活的NLRP3炎性体剪接IL-1β前体以进行成熟和分泌,并切割GSDMD以引发焦亡。通过Western blot和ELISA评估,TNF-Exo+ATP组或nigericin组的GSDMD n端和IL-1β的分泌增加(图2f-g)。此外,通过RT-qPCR测定,TNF-Exo增加巨噬细胞中NLRP3和caspase-1 mRNA的表达(图2h-i)。这些数据表明,TNF-Exo仅作为上调NLRP3炎性体表达的启动信号,而第二个信号,如ATP或nigericin,则需要在体外最终诱导巨噬细胞焦亡。
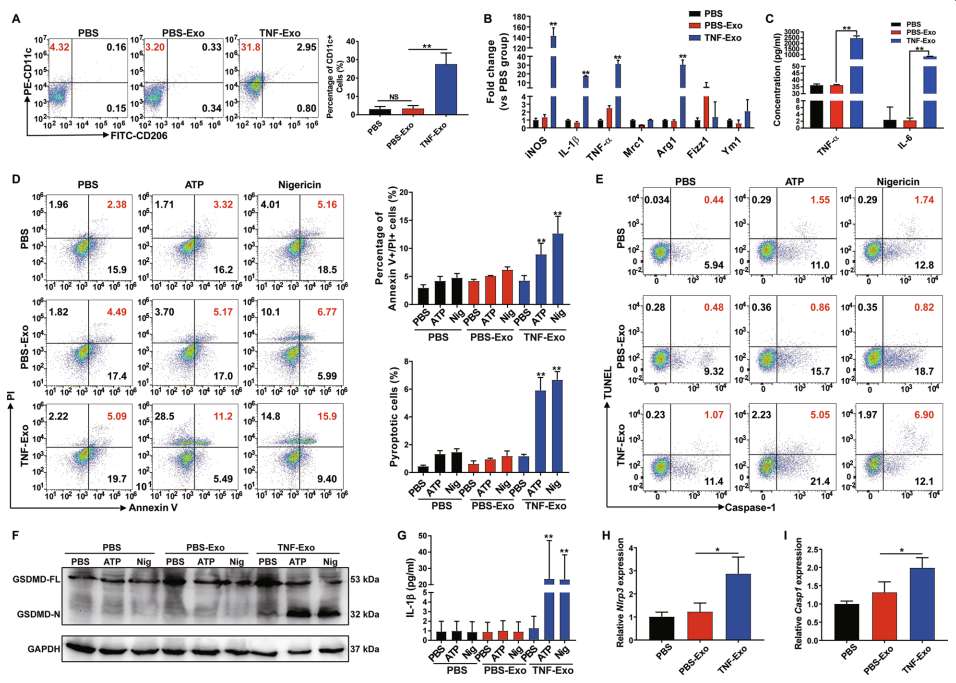
图2体外共培养模型中,TNF-Exo通过NF-κB途径促进M1巨噬细胞激活并引发巨噬细胞焦亡
3. PMN来源外泌体的miRNA分析
了解脓毒症休克患者的外泌体可以传递致病途径相关的miRNAs,这可能是脓毒症期间细胞间通信的一种新机制。随后对pmn来源的外泌体进行了miRNAs筛选,检测到26个miRNAs。研究发现,与PBS-Exo相比,TNF-Exo的表达量增加了两倍(图3a)。富集通路分析也对这26个miRNAs进行了信号转导相关最富集的通路分析,数据显示NF-κB信号通路在20条最富集的通路中(图3b)。通过查阅文献,发现miR- 30d-5p的表达与NF-κB信号通路呈正相关,且RT-qPCR发现miR-30d-5p在TNF-Exo中的表达明显高于PBS-Exo(图3c)。TNF-α刺激后PMNs中的miR-30d-5p水平下降,这表明miR-30d-5p从细胞室转移到外泌体(图3d)。用TNF-Exo体外培养的巨噬细胞显示出更高水平的miR-30d-5p(图3e)。这些结果表明,TNF-α可以增强miR-30d-5p在PMN外泌体中的装载并转移到受体巨噬细胞中。因此,推测TNF-Exo可能将miR- 30d-5p转移到巨噬细胞中,进而激活NF-κ b信号通路。
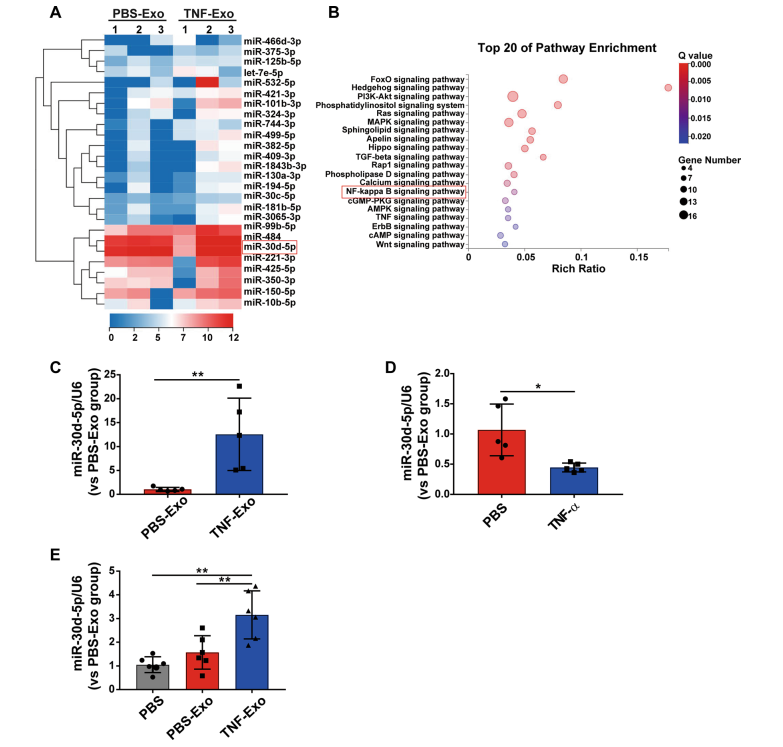
图3 PMN来源外泌体的miRNA分析
4. TNF-Exo通过miR-30d-5p激活巨噬细胞NF-κB信号通路
为了验证上述假设,在与TNF-Exo培养之前,用miR-30d-5p抑制剂转染Raw264.7巨噬细胞。发现转染miR-30d-5p抑制剂可逆转由TNF-Exo诱导的M1巨噬细胞标记物和促炎细胞因子的上调(图4a-c)。此外,抑制miR-30d-5p显著降低了TNF-Exo处理的受体巨噬细胞中NF-κB p-p65蛋白的表达(图4d)。此外,在用TNF-Exo培养之前,用miR-30d-5p抑制剂处理Raw264.7巨噬细胞可降低NLRP3和caspase-1的mRNA水平(图4e)。转染miR-30d-5p抑制剂对TNF-Exo加ATP的caspase-1激活有显著的抑制作用(图4f-g)。TNF-Exo+ATP刺激上调的GSDMD n端也被miR-30d-5p抑制剂抑制(图4h)。这些数据表明,外泌体miR-30d-5p通过激活NF-κB信号通路促进M1巨噬细胞活化,启动巨噬细胞焦亡。

图4 TNF-Exo通过miR-30d-5p激活巨噬细胞NF-κB信号通路
5. 外泌体miR-30d-5p通过靶向SOCS-1和SIRT1激活巨噬细胞NF-κB
生物信息学分析表明,细胞因子信号抑制因子SOCS-1和sirtuin 1可能是miR-30d-5p的靶基因,也是NF-κB信号通路的负调控因子。通过Targetscan预测的那样,miR-30d-5p可能保留了SOCS-1和SIRT1的3’-UTR的结合位点(图5a)。双荧光素酶报告分析,发现SOCS-1/SIRT1 3’-UTR WT组中的miR-30d-5p过度表达显著降低了荧光素酶活性,但3’-UTR Mut组中没有(图5b)。在Raw264.7巨噬细胞中,miR-30d-5p过表达抑制了SOCS-1和SIRT1的mRNA和蛋白水平(图5c-d)。所有这些结果都表明SOCS-1和SIRT1是miR-30d-5p的直接靶基因。为了确定外体miR-30d-5p是否靶向巨噬细胞中的SOCS-1/SIRT1,研究发现TNF外泌体处理的巨噬细胞中SOCS-1和SIRT1的mRNA和蛋白质表达均降低(图5e–f),而miR-30d-5p抑制剂逆转了SOCS-1和SIRT1的表达(图5g)。研究表明SIRT1通过降低NF-κB p65亚基赖氨酸310的乙酰化水平来降低NF-κB活性。在这个研究中TNF-Exo后p65赖氨酸310乙酰化增强(图5f),而抑制miR-30d-5p后p65赖氨酸310乙酰化减弱(图5g)。这些结果表明,外泌体miR-30d-5p靶向巨噬细胞中的SOCS-1和SIRT1,随后通过增加p65赖氨酸310的乙酰化作用激活NF-κB。

图5外泌体miR-30d-5p通过靶向SOCS-1和SIRT1激活巨噬细胞NF-κB
6. 抑制miR-30d-5p可减轻TNF Exo或CLP诱导的肺损伤
在体内研究miR-30d-5p在TNF-Exo中的功能作用。发现注射TNF Exo后,肺组织中的miR-30d-5p表达显著增加(图6a)。在注射TNF-Exo之前,通过小鼠尾静脉给予miR-30d-5p抑制剂或置位阴性对照,并检测肺组织中促炎细胞因子和NLRP3炎性体的表达水平。结果显示,在TNF-Exo给药后,miR-30d-5p抑制剂显著降低了IL-1β、iNOS、NLRP3、caspase-1mRNA的表达水平(图6b-c)。此外,抑制miR-30d-5p可降低TNF-Exo诱导的肺中M1巨噬细胞活化和巨噬细胞死亡(图6d,e)以及组织学损伤(图6f)。
此外,应用CLP小鼠模型模拟脓毒症,进一步证实miR-30d-5p在体内的作用。CLP小鼠肺组织中miR-30d-5p的表达也显著增加(图6g)。在miR-30d-5p抑制CLP模型中,IL-6、iNOS、NLRP3 mRNA表达下降(图6h,i),M1巨噬细胞活化和肺内巨噬细胞死亡减少(图6j,k)以及肺损伤减轻(图6l)。值得注意的是,miR-30d-5p抑制的CLP小鼠的存活率显著高于未抑制的CLP小鼠(图6m)。所有这些数据表明,抑制miR-30d-5p可通过抑制M1巨噬细胞活化和巨噬细胞死亡,从而改善TNF-Exo和CLP诱导的肺损伤。
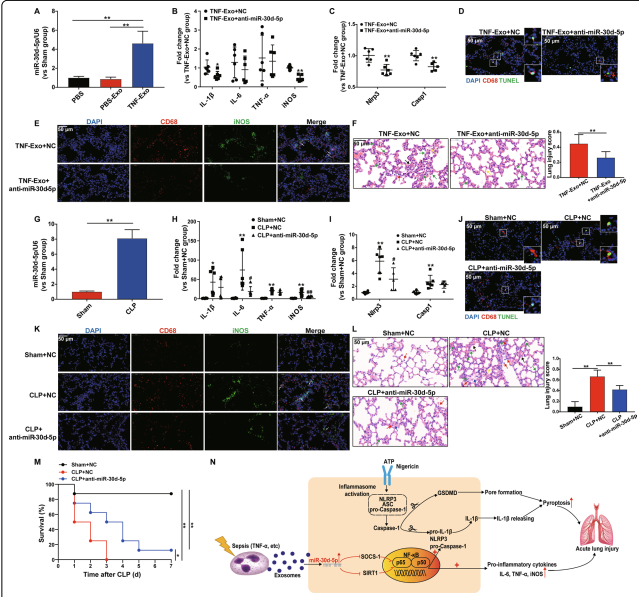
图6 miR-30d-5p抑制减轻TNF-Exo或CLP诱导的肺损伤
主要结论:
本研究表明,PMNs来源的外泌体miR-30d-5p可以通过增强M1巨噬细胞极化和启动巨噬细胞焦亡促进肺部炎症。调节嗜中性粒细胞和巨噬细胞之间的交互作用,可减轻脓毒症相关ALI期间的组织炎症和损伤,突出了其作为脓毒症相关ARDS治疗策略的潜力(图6n)。
