






ADAR1限制ZBP1介导的免疫应答和PANoptosis以促进肿瘤发生
细胞死亡提供宿主防御并维持体内平衡。Z-DNA结合蛋白1 (ZBP1)激活炎症性细胞死亡(PANoptosis),而作用于RNA 1的腺苷脱氨酶(ADAR1)作为RNA编辑器维持体内平衡。最新研究结果表明,ADAR1抑制ZBP1介导的PANoptosis,促进肿瘤的发生。明确ADAR1和ZBP1在细胞死亡中的功能对于制定癌症和其他疾病的治疗策略至关重要。该研究发表2021年10月发表在《Cell Reports》,IF:9.423。
技术路线:
主要研究结果:
1. IFN信号转导增强核转运抑制剂诱导的细胞死亡
前期大量文献证明了NEIs诱导的细胞死亡途径对于理解NEIs抑制肿瘤发生的分子机制至关重要。为了确定NEIs是否能诱导细胞死亡,用NEIs KPT-330或LMB治疗骨髓来源的巨噬细胞(BMDM)。KPT-330或LMB治疗24小时后,BMDM中的细胞死亡水平较低(图1A和1C)。而与单独使用KPT-330或LMB治疗相比,IFN-b或IFN-g联合KPT-330或LMB治疗增加了细胞死亡的发生率(图1A- 1C),这表明IFN信号增强了NEIs诱导的细胞死亡。
另外,用NEIs处理可产生少量活性GSDMD P30片段,仅对NEIs反应时,caspase-1和caspase-11的切割量最小,与产生的GSDMD P30的数量一致(图1D)。除了pyroptosis,还发现KPT-330或LMB诱导BMDM中凋亡效应物的激活,如凋亡的caspase-8、-3和-7的裂解(图1E)。用KPT-330或LMB刺激的细胞显示MLKL的低水平磷酸化(图1F),表明坏死效应物的激活正在发生。
与细胞死亡的发生率一致,IFN-b或IFN-g治疗可增强KPT-330-或LMB诱导的caspase-1和GSDME的裂解(pyroptosis);caspase-8、-3和-7的裂解(apoptosis);和MLKL磷酸化(necroptosis)(图1D-1F)。总的来说,这些数据表明,IFN与NEIs联合使用可使细胞发生炎性小体激活和细胞死亡,包括焦亡、凋亡和坏死,表明PANoptosis正在发生。
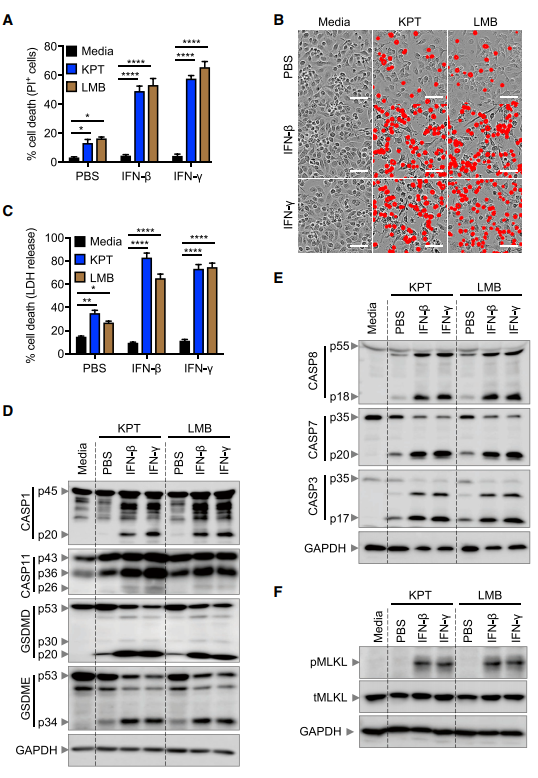
图1. 干扰素增强核输出抑制剂诱导的细胞死亡
2. ZBP1参与RIPK3信号通路激活NLRP3炎症小体和NEIs诱导的PANoptosis
在IFN-b和NEI治疗后,与野生型(WT)BMDM相比,缺乏ZBP1的BMDM减少了细胞死亡(图2A、2C)。与这种保护作用相一致,Zbp1–/– BMDMs显示pyroptotic, apoptotic, 和 necroptotic分子的激活减少(图2D-2F),表明Zbp1是IFN-和NEI诱导的NLRP3炎症小体激活和PANoptosis所必需的。此外,使用IFN-b和KPT-330或IFN-b和LMB治疗后,结果与IFN-b和NEI治疗后一致。

图2. ZBP1在IFN和核出口抑制剂的联合作用下触发炎症小体激活和细胞死亡
3. ADAR1抑制ZBP1介导的NLRP3炎症小体激活和PANoptosis
从髓系细胞中缺乏ADAR1的小鼠(ADAR1 / BMDMs)中获得BMDMs,以研究ADAR1在NEI介导的炎症小体激活和PANoptosis中的作用。与WT和Zbp1–/–细胞相比,缺乏ADAR1的细胞中IFN-b和KPT-330或IFN-b和LMB联合诱导的细胞死亡增加(图3A和3B)。此外,与WT和Zbp1–/–BMDM相比,缺乏ADAR1的BMDM加速了细胞死亡(图3A)。尽管ADAR1-p150和ZBP1都是IFN可诱导且包含Za结构域,这些分子在细胞死亡方面表现出不同的表型:ZBP1的丢失抑制细胞死亡,ADAR1的丢失促进细胞死亡(图2、3A和3B)。
单独使用KRT -330或LMB治疗后,Adar1 / BMDMs细胞死亡增加(图3C和3D)。Adar1–/– BMDMs增加了caspase-1的裂解和pyroptotic分子GSDME的活化;apoptotic的caspase;以及单独使用KRT -330或LMB处理后的坏死分子MLKL(图3E-3G),提示ADAR1抑制NEIs诱导的NLRP3炎症小体激活和PANoptosis。同时缺乏ADAR1和ZBP1的BMDMs在KPT-330或LMB作用下的细胞死亡与WT细胞相似(图4A和4B)。此外,NEIs处理的Adar1–/– BMDMs中,ZBP1的缺失抑制了NLRP3炎症小体的激活以及焦亡、凋亡和坏死效应物的激活(图4C-4E)。
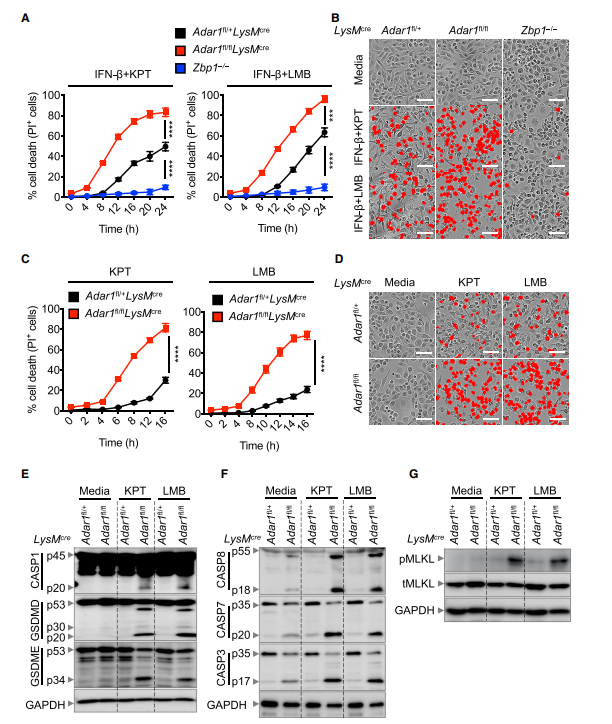
图3. ADAR1缺陷导致核输出抑制剂导致细胞加速死亡

图4. 缺失ZBP1拯救ADAR1缺陷细胞中核输出抑制剂诱导的炎症体激活和细胞死亡
4. IFNs促进NEIs产生EREs并激活ZBP1
经KPT-330或LMB处理后,Adar1–/– BMDMs中的dsRNA数量增加(图5A和5B)。为了测试KPT-330或LMB处理是否能诱导ERE衍生的dsRNA,结果显示用KPT-330或LMB处理Adar1–/– BMDM可增加ERE的表达(图5C)。与培养基处理的WT细胞相比,单独使用KPT-330或LMB处理的WT细胞增加了dsRNA水平(图5D)。然而,与PBS、KPT-330或LMB处理的WT细胞相比,IFN-b和KPT-330或IFN-b和LMB的组合更有力地增加了dsRNA(图5D和5E)。此外,单独使用KPT-330或LMB治疗的细胞中,ERE的表达上调,在联合使用IFN-b和KPT-330或IFN-b和LMB治疗的细胞中更显著地增加(图5F)。
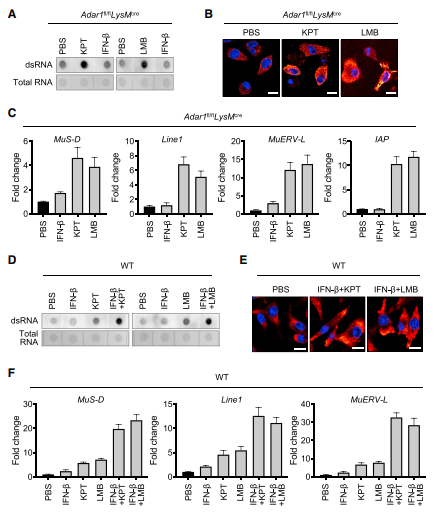
图5. IFN信号通过核出口抑制剂增强双链RNA (dsRNA)的积累
5. ADAR1与RIPK3竞争ZBP1结合抑制炎症小体激活和细胞死亡
在IFN-b刺激下,ADAR1和ZBP1的表达上调(图6A)。此外,LMB治疗能够诱导ADAR1和ZBP1的表达,但不如IFN-b刺激那么强烈(图6A)。在基础条件下未观察到ZBP1和ADAR1之间的相互作用(图6B)。然而,在IFN-b刺激的WT细胞中,观察到ZBP1与ADAR1的强烈相互作用,而不是与RIPK3的强烈相互作用(图6B)。虽然单独使用KPT-330或LMB处理的WT细胞显示出ZBP1与ADAR1和RIPK3都有一定的相互作用,但IFN-b与KPT-330或LMB联合刺激则减少了ZBP1与ADAR1的相互作用,增加了ZBP1与RIPK3的相互作用(图6B)。
WT ZBP1与ADAR1共免疫沉淀显示ZBP1与RHIM或C末端结构域相互作用(图6C)。在WT BMDM中未检测到ADAR1和ZBP1或RIPK3和ZBP1之间的任何相互作用(图6D)。经IFN-b和KPT-330或IFN-b和LMB治疗后,ZBP1与WT BMDM中的ADAR1和RIPK3相互作用(图6B和6D)。然而,在缺乏ADAR1的情况下,ZBP1和RIPK3之间的相互作用增加(图6D和S6D)。此外,与WT细胞相比,Ripk3–/–细胞中ZBP1和ADAR1之间的相互作用增加(图6D),表明ADAR1与Ripk3竞争结合ZBP1。此外,在缺乏其Za2结构域的情况下,在经IFN-b和KPT-330治疗的BMDM中,ZBP1没有与RIPK3相互作用(图S6E),这表明ZBP1的Za2结构域是促进ZBP1和RIPK3的RHIM结构域之间相互作用所必需的。在BMDMs中沉默PKR,并使用IFN-b与KPT-330或LMB联合治疗,观察到细胞死亡没有差异(图S6F),表明这是一个PKR独立的机制。

图6. ADAR1与RIPK3竞争与ZBP1的结合
6. ADAR1通过抑制ZBP1介导的炎症细胞死亡促进肿瘤发生
与同窝对照小鼠相比,Adar1fl/flLysMcre小鼠结肠的肿瘤负担在肿瘤数量和肿瘤大小方面都较低(图7A-7C)。Adar1fl/flLysMcreZbp1–/–小鼠显示出与对照小鼠相似的肿瘤负担(图7D 7F)。此外,ZBP1 Za2结构域的缺失产生了类似的结果(图7D-7F),表明ZBP1 Za2结构域在抑制骨髓细胞中缺乏ADAR1的小鼠的结直肠肿瘤发生中至关重要。
关于IFN和NEIs治疗黑色素瘤的疗效方面。B16黑色素瘤细胞植入后8天用KPT-330治疗的WT小鼠显示黑色素瘤生长减少(图7G和7H)。此外,IFN-g和KPT-330联合治疗的小鼠肿瘤消退显著改善(图7G和7H)。为了确定肿瘤消退是否依赖于ZBP1,将IFN-g和KPT-330联合应用于WT和ZBP1/小鼠。IFN-g和KPT-330未能使Zbp1–/–小鼠中的黑色素瘤消退(图7I)。ZBP1的Za2结构域的缺失产生了类似的结果(图7I),表明ZBP1,尤其是其Za2结构域,对于抑制IFN和NEIs联合治疗的肿瘤发生至关重要。
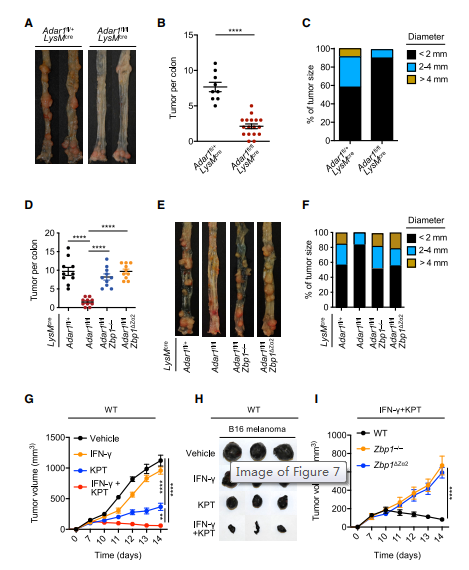
图7. ADAR1-ZBP1相互作用的调节影响肿瘤的发生
主要结论:
这项研究结果表明ADAR1通过抑制ZBP1介导的炎症细胞死亡和PANoptosis,作为一个限制抗肿瘤免疫的检查点,并提示影响ADAR1-ZBP1相互作用的策略,如使用IFN和NEIs治疗可能是有前景的。这些结果提供了更深入的理解细胞死亡的细胞和分子机制,特别是PANoptosis,并将为发展有效的治疗策略对抗癌症提供重要的信息。
参考文献:
Karki R, Sundaram B, Sharma BR, Lee S, Malireddi RKS, Nguyen LN, Christgen S, Zheng M, Wang Y, Samir P, Neale G, Vogel P, Kanneganti TD. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep. 2021; 37(3):109858. doi: 10.1016/j.celrep.2021.109858. PMID: 34686350.