






LIFR−NF-κB−LCN2轴控制肝脏肿瘤的发生和对铁死亡的易感性
肝细胞癌(hepatocellular carcinoma,HCC)是肝细胞中最常见的实体肿瘤,严重威胁患者的生命健康。铁死亡在癌症中具有较大的治疗潜力。本研究探讨高甲基化水平基因LIFr在调控HCC细胞铁死亡中发挥的作用。本文于2021年12月发表在《Nature communications》杂志上,IF=12。298。
本研究技术路线:

本研究主要结果:
1. LIFR在HCC中下调,LIFR的缺失促进肝癌的发生
基于TCGA数据库的测序数据和qPCR分析显示 ,与正常组织相比,LIFR mRNA水平在HCC中显著下调 (Fig 1a–c)。接下来作者通过n -亚硝基二乙胺(DEN)诱导小鼠肝脏肿瘤。 5只一岁的DEN处理的C57BL/6小鼠中有4只肝脏肿瘤中Lifr的表达低于正常小鼠肝脏组织 (Fig 1d)。
为了研究LIFR的功能,在 C57BL/6 菌株通过 LoxP-flanked (floxed)处理获得 Lifr flox/flox(Lifr fl/fl )纯合子。然后,作者通过 albumin-Cr小鼠获得肝细胞特异性Lifr缺失突变体(Lifr fl/ fl; Alb-Cre)小鼠。与预期一样,Alb-Cre小鼠肝脏中Lifr mRNA和蛋白水平下降 (Fig 1e-f)。为了确定肝脏表型,作者跟踪了一组小鼠到 2岁。结果发现在14只Lifr fl/ flAlb-Cre小鼠中有4只观察到肉眼可见的肝脏肿瘤(4只小鼠的肿瘤数量分别为1、2、3和4),而12只Lifr fl/fl小鼠均未显示可见肿瘤(Fig 1g)。与此同时,14日龄时,作者对Alb-Cre小鼠腹腔注射DEN。并在7个月后对所有小鼠实施安乐死,结果发现在20只Lifr fl/flAlb-Cre小鼠中检测到4只存在显著的HCC肿瘤负担,但在15只Lifr fl/fl对照小鼠中没有发现 (Fig 1h)。Alb-Cre小鼠从第5个月时开始死于肝脏肿瘤,Lifr fl/fl小鼠从第14个月时开始死于肝脏肿瘤。综上所述,这些数据表明Lifr是自发和致癌诱导的肝脏肿瘤的抑制因子。此外,LifrLifr的缺失显著加重了癌基因诱导的肝细胞瘤,并增加了肝脏重量和肝脏体重比(Fig 1j-m)。
为了解成年小鼠中Lifr缺失的影响。枸橼酸他莫昔芬注射到小鼠中处理5天之后进行观察,发现Lifr fl/fl ;Cre-ERT2小鼠和Lifr fl/fl小鼠肝癌明显恶化(Fig 1n-q),表明 LIFR 是成年肝癌基因诱导的抑制因子。

Fig 1 Lifr的缺失会促进肝癌的发生
2. LIFR对诱导铁死亡的药物具有敏感性
为了评估LIFR的表达是否与药物反应相关,作者使用了癌症治疗反应门户(CTRP)分析了基因表达与癌症细胞系中481种化合物的反应之间的相关性。来自CTRP的肝癌细胞系数据显示LIFR表达和铁死亡诱导剂敏感性密切相关 (Fig 2a)。为了确定LIFR是否能调节铁死亡,作者用铁死亡诱导物和另外两种广泛使用的铁死亡诱导物RSL3和胱氨酸刺激肝细胞。结果发现这三种处理都触发了对照组PHM细胞的大量细胞死亡;相比之下,敲除LIFR的PHM细胞对诱导细胞凋亡具有抗性(Fig 2b)。相反,过表达LIFR可使PHM细胞对所有三种铁诱导物敏感(Fig 2c)。
最近,相关研究报道sorafeni在某些条件下可以触发铁死亡。有趣的是,在sorafeni治疗后,细胞凋亡或坏死抑制剂(而不是凋亡或坏死抑制剂)可以在体外恢复肝癌细胞活力。这促使作者研究LIFR是否调节索拉非尼的敏感性。对小鼠进行为期一周的sorafeni处理后发现,,索拉非尼治疗大大提高了LIFRfl/fl 小鼠存活率(Fig 2d)。索拉非尼治疗对LIFRfl/fl 小鼠具有较好的抗肿瘤作用而对LIFRfl/fl Alb-Cre小鼠的作用不明显(Fig 2e)。
索拉非尼治疗后,与LIFRfl/fl Alb-Cre小鼠相比,LIFRfl/fl小鼠中4-HNE 的含量明显增加(Fig 2 f,g)。索拉非尼治疗可使 HCC 肿瘤中4- HNE水平升高(Fig 2j,k) ,而 LIFR 过表达可使4- HNE水平进一步升高,而lipro-1联合治疗可逆转4-HNE 水平(Fig 2j,k)。这些数据表明 LIFR 抑制HCC 的生长并提高对索拉非尼诱导的铁死亡敏感性。
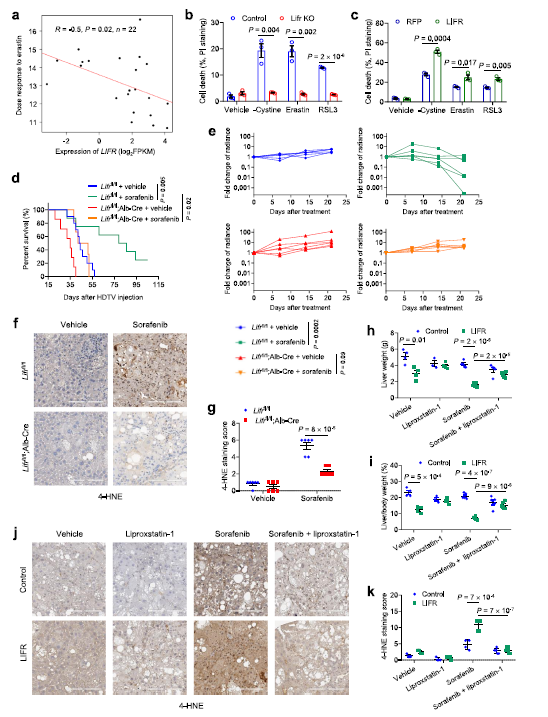
3. LIFR是NF-κB信号通路和LCN2的负调控因子
为了确定Lifr丢失对肝脏转录组的影响,作者对Lifrfl/fl和Lifrfl/fl;Alb-Cre小鼠肝脏组织进行了RNA-seq分析。Lcn2是Lifrfl/fl;Alb-Cre小鼠肝脏中上调倍数最高的基因之一,已有研究报道Lcn2促进乳腺肿瘤的发生和转移,其在肝癌中表达上调。由于Lcn2具有胞质和分泌形式,可以由中性粒细胞和肝细胞分泌,于是作者分析了对照细胞和 LIFR 基因敲除后 PHM 细胞分泌的细胞因子,发现 LCN2是 LIFR 丧失后前五位上调的细胞因子之一(Fig 3b)。进一步通过 qPCR (Fig 3 c)和 ELISA (Fig 3 d)验证了 LCN2在LIFR 敲除 PHM 细胞中的表达,以及 LCN2在LIFRfl/fl、 Alb-Cre 小鼠血清中的表达(Fig 3 e)。LIFR 基因敲除后 PHM 细胞p65磷酸化水平显著升高,LIFR 基因重新表达可逆转 p65磷酸化水平(Fig 3f) ,在 LIFR 敲除的肝癌细胞系 Mahlavu 和 PLC/PRF/5中观察到同样的效果(Fig 3g -f)。
与lifr缺失的PHM cell相似(Fig 3f),lifr敲除的小鼠肝脏中p65磷酸化(Fig 3i, j)以及Lcn2 mRNA(Fig 3k, l)和蛋白水平均上调(Fig 3m, n),表明NF-κB信号通路的激活。与此相反, Stat3、 Yap、 Akt 和 Erk 的磷酸化水平并没有显著下降(Fig 3i,j)。此外,他莫昔芬诱导 LIFR 上调p65磷酸化,但不影响肝内 stat3磷酸化(Fig 3o)。Lcn2在LIFR过表达的PHM细胞中下调,在癌基因诱导的HCC模型中,腺病毒介导的LIFR降低了肝组织中p65磷酸化和Lcn2蛋白水平(Fig 3p, q),并降低了血清中的Lcn2水平。

Fig 3 LIFR负向调节NF-κB信号通路和肝脏LCN2
4 . LIFR的缺失通过SHP1激活NF-κB信号通路,导致LCN2的上调
为了了解LIFR如何调节NF-κB信号,作者通过一个蛋白质相互作用数据库获取LIFR潜在的相互作用蛋白。在所有候选基因中,磷酸酶SHP1下调肝癌细胞 p65磷酸化已被报道。结果发现,SHP1,而非SHP2,被SFB 标记的LIFR 蛋白通过 s 蛋白珠拉下来,但是没有被 SFB标记的 GFP 蛋白拉下来(Fig 4 a,b)。SHP1已经被证明可以与TRAF6相互作用,并抑制K63-linked的泛素化,导致NF-κB信号通路失活。而E3连接酶TRAF6介导k63连接的泛素化和TAK1的激酶激活,进而在NF-κB通路中激活IKK。作者过表达LIFR降低了 TRAF6的 k63连锁泛素化和 p65的磷酸化,这可能是SHP1的降低导致的(Fig 4c),这表明LIFR 通过 SHP1抑制TRAF6的泛素化和 NF-κB 信号转导。LIFR 在人肝癌细胞系 PLC/PRF/5中的过表达导致 IKKα/β 磷酸化水平下调,IκBα 蛋白水平上调,SHP的降低可消除这些影响(Fig 4d)。此外,LIFR 的降低显著增加 p65磷酸化,减弱 SHP1和traf6之间的相互作用,而不影响SHP1564位点的磷酸化(SHP1磷酸酶活性的指标)(Fig 4e)。这些数据表明 LIFR 促进SHP1- TRAF6的相互作用,进而抑制 TRAF6泛素化和 NF-κB 信号通路。
为探讨NF-κB是否介导LIFR缺失的HEK293T、PLC/PRF/5和PHM细胞株中LCN2的上调,作者使用shRNA敲除了这3个细胞系中的p65,结果发现p65的缺失逆转了LCN2诱导的LIFR降低(Fig 4f–h)。肝细胞特异性敲除LIFR 显著增加了肝组织中 LCN2的表达水平,而体内敲除 p65或 LCN2可逆转这一过程(Fig 4 i,j)。p65 和LCN2的干扰阻断了肝细胞特异性LIFR基因敲除所诱导的肝癌发生(Fig 4k-m),提示LIFR 基因的缺失一定程度上能通过 NF-κB 和 LCN2促进肝癌的发生。

Fig 4 LIFR的缺失通过SHP1激活NF-κB信号通路,导致LCN2的上调
5. LIFR和SHP1对铁死亡正向调控,LCN2对铁死亡负向调控
作者发现,用铁死亡诱导剂处理HT1080细胞一段时间后会诱导细胞死亡,而敲除LIFR或SHP1可以在铁死亡诱导剂处理5和10小时后保护HT1080细胞免受细胞死亡 (Fig 5a-d)。此外,LIFR 或 SHP1基因敲除可以减少RSL3、 fin56或 fino2诱导的细胞死亡(Fig 5e,f)。另一方面,随着时间的推移,LCN2的下调使HT1080细胞敏感,铁死亡诱导剂诱导的细胞死亡,可以通过与铁死亡抑制剂liproxstatin-或铁螯合剂 deoxamine (DFO)的联合治疗来挽救 (Fig 5g)。在用 RSL3,fin56或 fino2处理的ht1080细胞时观察到类似的效果(Fig 5h)。敲低LIFR 或 SHP1可降低脂质过氧化(Fig 5i,j),而敲低 LCN2可升高脂质过氧化,这中效果能被 liprostatin-1或 DFO 所逆转(Fig 5k)。这些数据进一步证实 LIFR和 SHP1是铁死亡的正向调节因子,而 LCN2是铁死亡的负向调节因子。
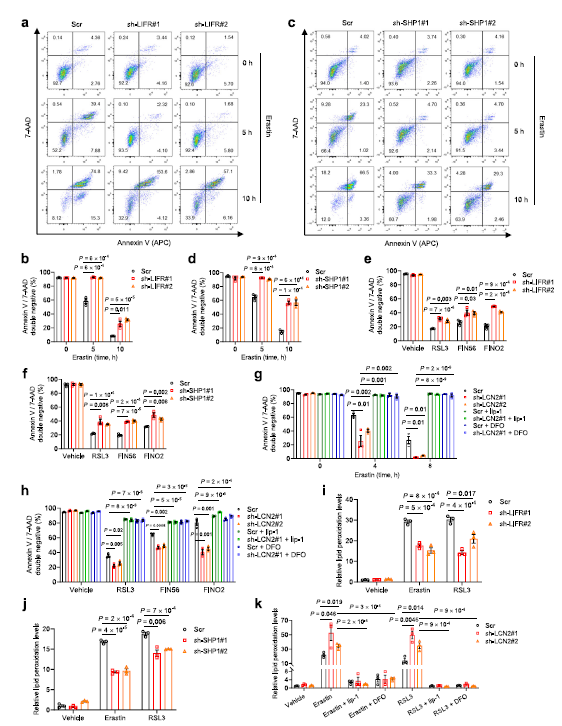
Fig 5 LIF和SHP1对铁死亡正向调控,LCN2对铁死亡负向调控
6. LCN2介导铁死亡耐药,是提高索拉非尼疗效的有效靶点
作者推测 LIFR 通过下调 LCN2调节铁死亡。实际上,LIFR过表达能提高PLC/PRF/5细胞对铁死亡诱导剂或索拉非尼的敏感度,这种效果可以通过与 liprostatin-1、 DFO 或纯化的 LCN2蛋白协同作用而逆转,这表明 LIFR 能通过LCN2使肝癌细胞对铁死亡诱导剂敏感。相反,在 Mahlavu 细胞中敲除LIFR,以及在PHM 细胞中敲除 LIFR,则上调LCN2导致的对铁死亡诱导剂和索拉非尼耐药性,而这种耐药性可以通过敲低LCN2而逆转(Fig 6 a,b)。
肝脏中LIFR缺失会引起铁(Fe2 +)水平下调(Fig 6c)。作者假设 lcn2的缺失是通过提高细胞中 Fe2 + 和脂质过氧化的水平来增加对铁死亡的敏感性。LIFR的缺失降低了细胞内 Fe2 + 水平和脂质过氧化水平,而 lcn2的敲除可以逆转这两种效应(Fig 6d,e)。另一方面,LIFR或 lcn2的缺失并没有改变谷胱甘肽(GSH)、 Slc7a11、 fsp1和 gpx4的水平(Fig 6f,g)。作者测量了LIFR敲除的PHM小鼠肝细胞系中铁的含量,结果发现LIFR的缺失降低了细胞中Fe2+和脂质过氧化的水平(Fig 6 d,e)。另一方面,LIFR或Lcn2的缺失并没有改变谷胱甘肽(GSH),Slc7a11, Fsp1和Gpx4的水平 (Fig 6f,g)。
为了进一步探索LCN2靶向制剂是否能提高索拉非尼的治疗效果,作者在 NSG小鼠中建立了4个 HCC 异种移植(PDX)模型。结果发现四个 PDX 株系(# 3-# 6)中有三个表现为 LIFR低表达,同时 LCN2和磷酸化 p65水平较高(Fig 6h)。在所有品系中,其中# 5株系LIFR 水平最低,lcn2水平最高,# 4株系则相反(Fig 6h)。在临床实验中,将#4和# 5 PDX中的PDX肿瘤组织植入NSG小鼠,当肿瘤达到50 – 100 mm3时开始治疗。将小鼠分为4个治疗组:(1)IgG + vehicle;(2) LCN2抗体+ vehicle;(3) IgG +索拉非尼;(4) LCN2抗体+索拉非尼。结果发现LCN2抗体+ vehicle以及LCN2抗体+索拉非尼组LCN2水平明显降低(Fig 6i)。无论是否与索拉非尼联合治疗,PDX # 4的小鼠中,LCN2联合抗体治疗不改变肿瘤的生长(Fig 6j,l)。在PDX # 5的小鼠中,单独使用LCN2联合抗体治疗对肿瘤生长的影响不大,而联合治疗比单独使用索拉非尼治疗具有更好的抗肿瘤效果(Fig 6k,l)。抗LCN2治疗并不影响增殖Ki-67和凋亡标记物caspase3的水平,而与索拉非尼联合治疗提高了4-HNE和 MDA(脂质过氧化的两个标记物)的水平(Fig 7a,b)。此外,电镜分析显示,在 PDX # 5,联合治疗组的肿瘤细胞中含有萎缩的线粒体和严重浓缩的膜,这是铁死亡典型的形态学特征。索拉非尼或LCN2抗体单独治疗也能增加线粒体膜密度,但程度要小于它们联合治疗(Fig 7c)。在 PDX # 4中,无论是单独使用索拉非尼治疗还是联合治疗都能诱导磷酸化铁蛋白相关的线粒体形态改变,而抗lcn2治疗并不能改变线粒体形态(Fig 7d)。总之,一种中和LCN2的抗体增强了索拉非尼对低LIFR表达和高LCN2表达的肝癌患者来源的异种移植瘤的铁诱导作用。
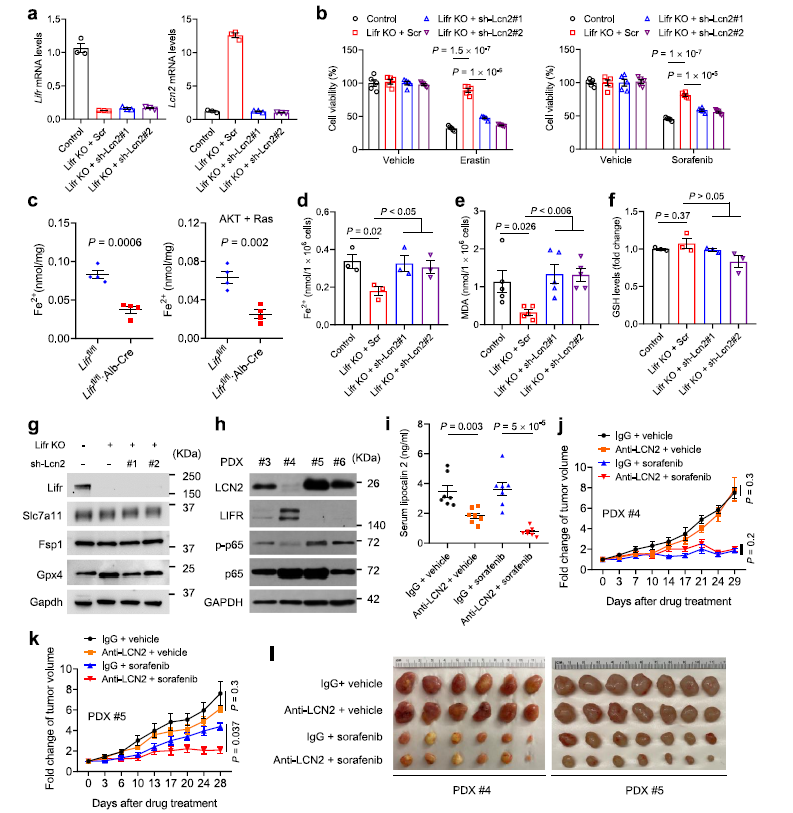
Fig 6 LCN2介导铁死亡耐药,是提高索拉非尼疗效的治疗靶点

Fig7一种中和LCN2的抗体增强了索拉非尼对低LIFR表达和高LCN2表达的肝癌患者来源的异种移植瘤的铁诱导作用。
LIFR 的缺失通过 SHP1 激活NF-κB 信号传导,导致铁螯合细胞因子 LCN2 的上调,导致对对铁死亡诱导剂不敏感。因此抗 LCN2 疗法可以通过靶向铁死亡来改善肝癌治疗。
参考文献:
[1] Yao Fan,Deng Yalan,Zhao Yang et al。 A targetable LIFR-NF-κB-LCN2 axis controls liver tumorigenesis and vulnerability to ferroptosis。[J] 。Nat Commun, 2021, 12: 7333。
