






浅谈DUSP26诱导主动脉瓣钙化的机制
钙化性主动脉瓣疾病(CAVD)的发病率和死亡率仍然很高,而治疗选择有限。在此,作者评估了双特异性磷酸酶26 (DUSP26)在CAVD中的作用和治疗价值。本研究于2021年6月发表在《European Heart Journal》IF:29.983期刊上。
技术路线:

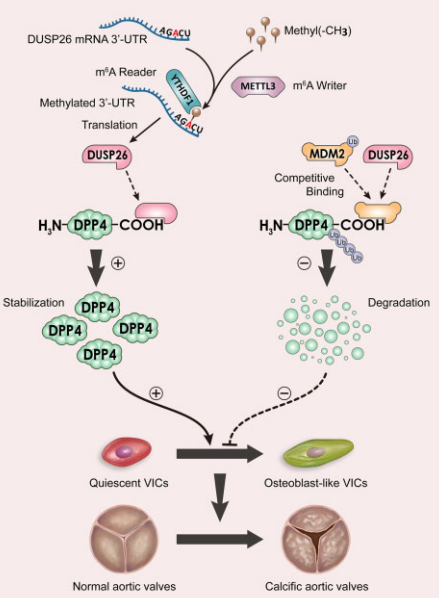
主要实验结果:
为了鉴定参与CAVD的基因,对人钙化性主动脉瓣(CAV)进行测序,获得差异表达基因谱(图1A)。预实验中,检测在CAV组最显著上调的10个差异表达基因在50对样本中的表达,有8个和测序结果的趋势一致,用shRNA干扰他们的表达检测,发现DUSP26对成骨标志基因的下调最显著,所以作者将DUSP26用于进一步实验。图1B-1D展示了DUSP26的表达,和成骨标志基因的表达。
为了评估m6A和DUSP26上调的相关性,作者分析了DUSP26序列,并在其接近终止密码子的3’-UTR处鉴定到一个METTL3保守序列(AGACU)(图1E-1F)。成骨刺激增加了原发性hVIC中m6A的整体水平,该作用分别被METTL3的沉默或过表达所减弱和加重(图1G)。RIP-qRT-PCR(图1H)和荧光素酶(图1I)证实了METTL3和DUSP26之间的相互作用。敲除METTL3显著降低了DUSP26蛋白的表达和半衰期,过表达则相反(图1J-1M)。这些结果表明METTL3介导的m6A修饰是CAVD中DUSP26表达增加的原因通过增加其稳定性。
METTL3通常以YTHDF1依赖的方式介导m4A修饰,所以探究了YTHDF1的作用。结果显示敲除YTHDF1显著降低了DUSP26的蛋白表达和半衰期,过表达则相反(图1N-1O)。YTHDF1和DUSP26的mRNA序列之间结合,这种结合作用可以被METTL3沉默和过表达所抑制或加强(图1P-1Q)。
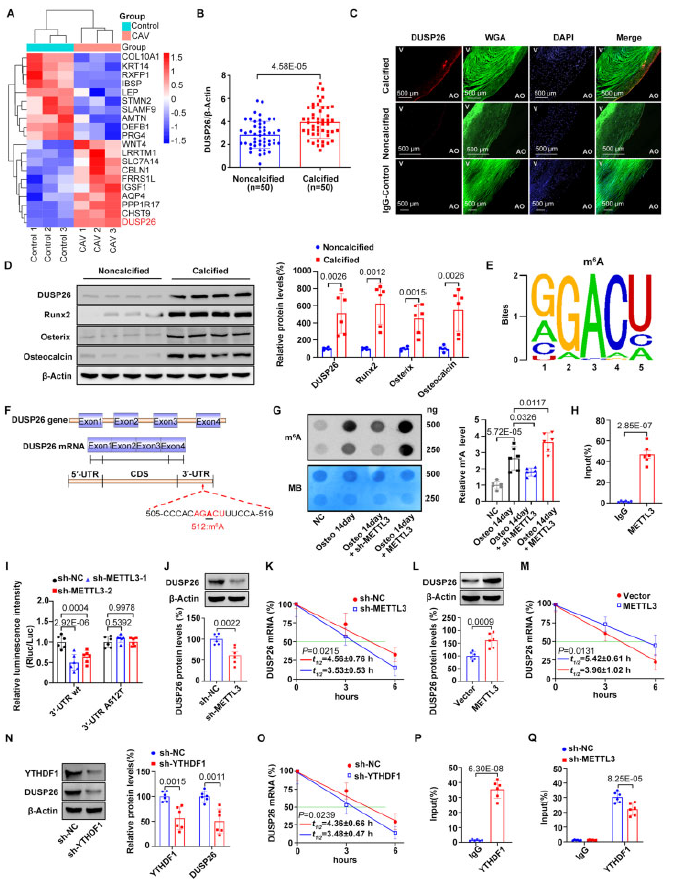
图1 DUSP26在人钙化主动脉瓣中上调,并经历METT3介导的m6A修饰
为了探究DUSP26在体内主动脉瓣钙化中的作用,作者在ApoE-/-小鼠中敲除DUSP26(AAV2-sh-DUSP26组;AAV2-scr为对照),24周后,HCD þ AAV2-scr组最大跨瓣喷射速度和平均跨瓣压力梯度显著升高,主动脉瓣面积明显减少;DUSP26的敲除可以部分回复这些变化(图2A-2C)。然后检测了ApoE-/-小鼠瓣叶的形态、纤维化和钙化。HCD促进主动脉瓣钙化,表现为主动脉瓣瓣叶厚度增加、胶原增加、钙离子沉积(图2D-2G)。而敲除DSP26可以部分恢复上述改变,并降低成骨标志物的表达。
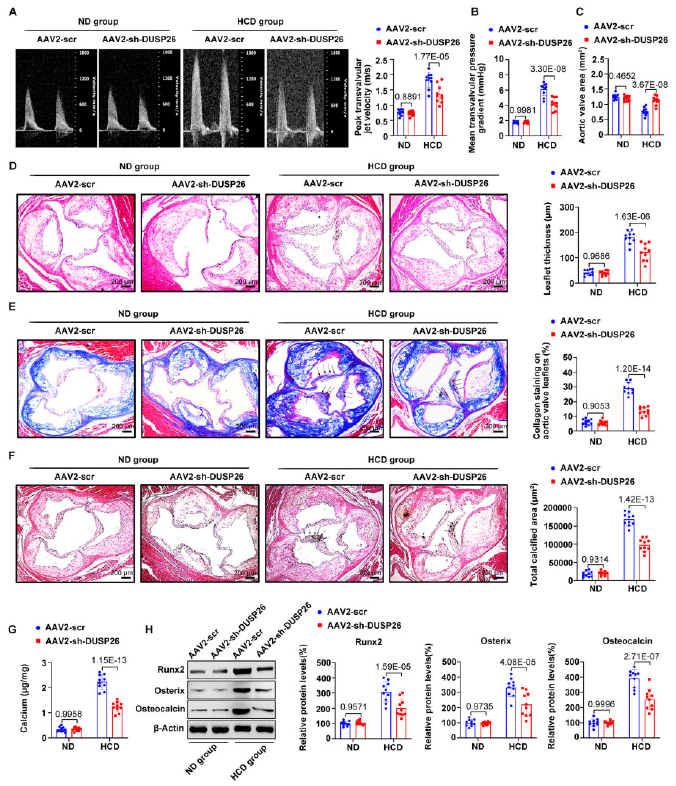
图2 DUSP26缺失可减轻体内主动脉瓣钙化
3、DUSP26敲除改善体外主动脉瓣钙化
作者从人主动脉瓣分离得到hVICs细胞用于体外实验。用成骨培养基诱导hVICs的成骨分化,发现DSP26的表达随着分化时间逐渐增加(图3A-3B)。随后敲除hVICs中的DUSP26,发现抑制了成骨分化标志基因的表达,以及钙化结节形成和钙沉积,过表达DUSP26的效果则相反(图3C-3D)。这些说明DUSP26沉默在体外也可改善主动脉瓣钙化。
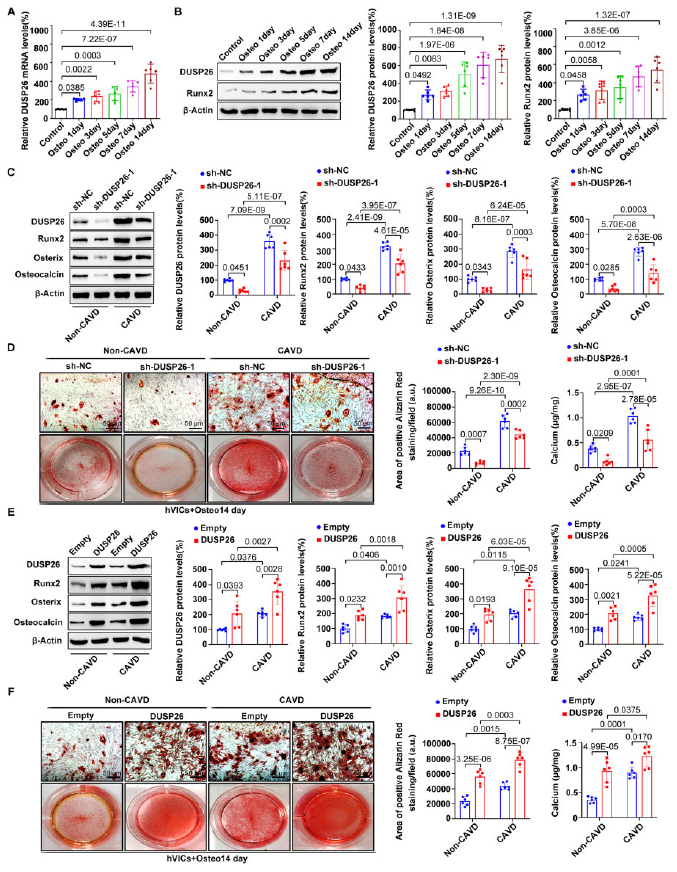
图3 DUSP26促进hVICs的成骨分化
4、在hVICs中,DUSP26和DPP4相互作用并抑制其降解
首先分离hVICs提取物并进行DUSP26抗体的免疫沉淀,进行MS分析,由此鉴定到DPP4是DUSP26的潜在结合蛋白(图4A-4B)。Co-IP实验验证两者间的结合关系(图4C-4D)。DUSP26是一个蛋白磷酸酶,于是探究其酶活性是否是两者结合所必须的。用F1063-0967处理或突变C152S抑制DUSP26的活性,结果发现其不影响两者间的结合(图4E-4F)。因此,DUSP26与DPP4相互作用不依赖于其磷酸酶活性。随后构建两个DPP4的截断性突变体,发现只有C端突变体可以和DUSP26结合(图4G-4H)。以上说明DUSP26和DPP4的C端结合。
随后探究DUSP26对DPP4的表达的影响。发现敲除DUSP26后DPP4的蛋白表达显著下降,过表达则相反,但都不影响mRNA的表达(图4I-4J)。DUSP26沉默诱导的DPP4下调被26S蛋白酶体抑制剂MG132减弱(图4K)。此外,DUSP26基因敲除缩短了hVIC中DPP4的半衰期,增加了DPP4的泛素化(图4L-4M)。
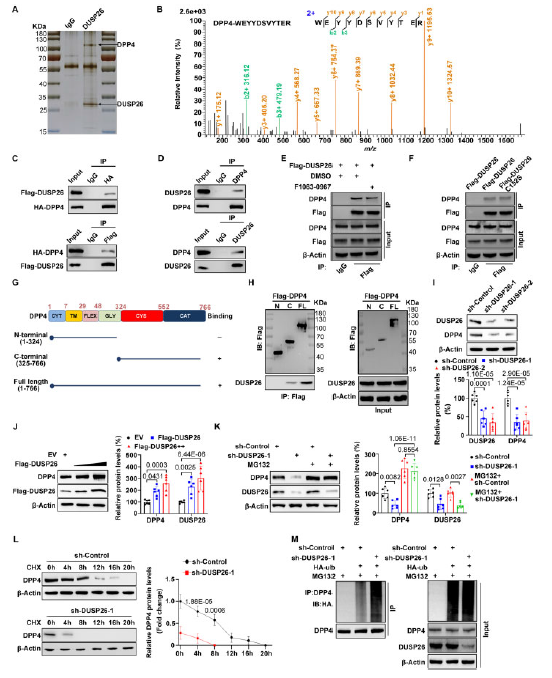
图4 DUSP26与DPP4相互作用,保护DPP4在hVICs中免受泛素介导的降解
5、DUSP26抑制hVICs中MDM2介导的DPP4的泛素化
作者在DPP4蛋白中发现了一个保守的MDM2结合基序(图5A)。为了探究MDM2是否作为DPP4的新型E3连接酶,通过co-IP来评估DPP4和MDM2之间的相互作用。在hVIC中异位表达的HA-DPP4与myc标记的MDM2共沉淀(图5B)。内源性DPP4和MDM2之间的相互作用也证实了(图5C)。此外,MDM2沉默后增加了DPP4的蛋白水平但对mRNA没影响(图5D)。相反,MDM2过表达降低了hVIC中DPP4的蛋白水平,这种作用受到MG132的抑制(图5E)。此外,MDM2过表达降低了hVIC中DPP4的半衰期(图5F),并增加了DPP4多聚泛素化(图5G)。这些数据表明DPP4确实是MDM2的底物。为了确定DPP4与MDM2结合的区域,进行了体外co-IP实验,发现DPP4的C端与MDM2相互作用(图5H),之前的结果中,DUSP26也和DPP4的C端作用,所以探索了DUSP26在hVIC中与MDM2结合DPP4竞争的能力。hVIC中DUSP26的沉默增强了MDM2和DPP4之间的相互作用(图5I)。相反,DUSP26过表达阻断了MDM2与DPP4的结合(图5J)。此外,DUSP26敲除降低了DPP4蛋白水平,在hVIC中,这种效应在DUSP26和MDM2共同敲低后减弱(图5K)。这些结果说明MDM2作为DPP4的新型E3连接酶,DUSP26与MDM2结合在DPP4的C端,从而提高DPP4蛋白在hVIC中的稳定性。

图5 DUSP26通过在hVIC中与MDM2介导的泛素化竞争来稳定DPP4
6、DPP4过表达挽救了hVIC中DUSP26沉默诱导的成骨分化表型
接下来,进行挽救实验来确定DUSP26是否通过DPP4的上调来发挥作用。DUSP26敲低的钙化减缓作用被DPP4过表达所逆转(图6A和6B)。相反,DUSP26过表达的促钙化作用被DPP4过表达所阻止(图6C和6D),表明DUSP26通过上调DPP4在hVICs中促进成骨分化。
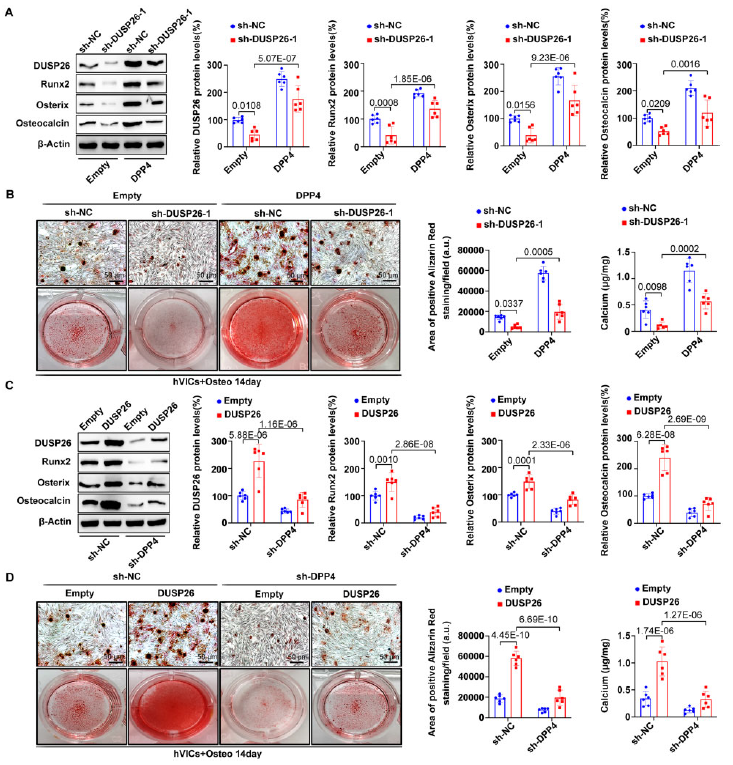
图6 DPP4过表达挽救了hVIC中DUSP26沉默诱导的成骨分化表型
参考文献:
Wang Yongjun., Han Dong., Zhou Tingwen., Chen Cheng., Cao Hong., Zhang Joe Z., Ma Ning., Liu Chun., Song Moshi., Shi Jiawei., Jin Xin., Cao Feng., Dong Nianguo.(2021). DUSP26 induces aortic valve calcification by antagonizing MDM2-mediated ubiquitination of DPP4 in human valvular interstitial cells. Eur Heart J, undefined(undefined), undefined. doi:10.1093/eurheartj/ehab316