






异戊基螺旋霉素I在核仁中靶向硒蛋白H进而抑制肿瘤和转移
与正常细胞相比,癌细胞表现出更高水平的氧化应激,这种更高水平的氧化应激也可以被利用来杀死肿瘤细胞,而使正常细胞完好无损。有研究发现Isovalerylspiramycin I (ISP I),一种新型大环内酯类抗生素,通过靶向核仁蛋白硒蛋白H (SELH),从而抑制癌细胞生长和肿瘤转移。该研究于2022年4月发表在《Journal of Experimental & Clinical Cancer Research》,IF:11.161。
技术路线:
主要研究结果:
1. Carrimycin(卡霉素)的结构与合成
卡霉素,主要由4 -O- (ISP) I、II、III和微量的4 -O-酰基螺旋霉素成分组成(图1A)。对产生卡里霉素的菌株进行基因组测序,发现了一个大约90 kb的生物合成基因簇 (图1B)。经过糖基化、氧化、酰基化和异戊基化等post-PKS修饰步骤后,ISP I、II和III的结果呈多相混合,差异仅在于碳3上羟基的酰基取代(图1C)。
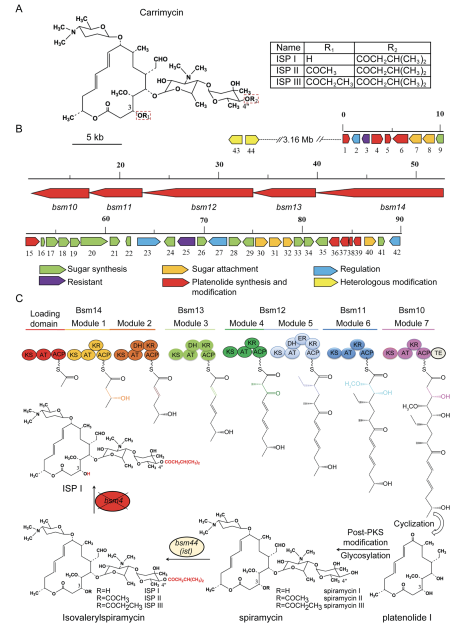
图1卡霉素生物合成基因簇的遗传结构及ISP I生物合成的主要步骤
2. ISP I抑制肿瘤的发生和转移
为了评估潜在的细胞毒性,用卡霉素主要成分ISP I、II和III治疗胶质母细胞瘤细胞株,并评估细胞活力:50%抑制浓度(IC50)值显示ISP I最有效,同时胶质母细胞瘤、肾细胞癌(RCC)和脑膜瘤在内的多种肿瘤细胞株对ISP的细胞毒性效应敏感(图2A和B)。流式分析显示,ISP I引起细胞周期阻滞和诱导剂量依赖性凋亡(图2C和D)。进一步用研究ISP I在体内的肿瘤抑制作用(图2E)。与对照组相比,使用ISP I的小鼠肿瘤生长显著降低(图2F和G)。另外,研究两种不同的、同基因的小鼠肺转移模型:黑色素瘤(B16)和乳腺癌(4T1)。C57BL/6小鼠静脉注射B16细胞,随机分为ISP处理组和盐水处理组(对照组)(图2H)。在两种肺转移模型中,ISP I显著降低了肺肿瘤负荷。ISP I治疗12天后,与盐水治疗的小鼠相比,B16小鼠肺转移的数量显著减少(图2I和J)。总的来说,这些发现表明,ISP I在体外抑制肿瘤生长。
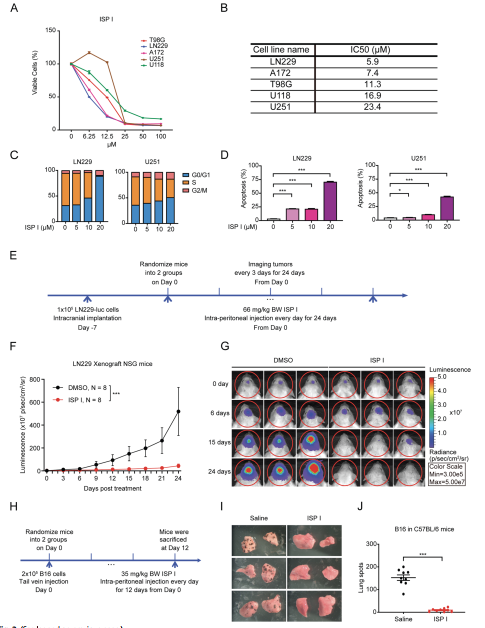
图2 ISP I抑制肿瘤生长,减少肺转移
3.ISP I以SELH为靶点
首先,在LN229细胞中进行了药物亲和反应靶稳定性(DARTS)检测(图3A显示了检测策略)。质谱分析显示SELH是ISP处理LN229细胞中最丰富的主要蛋白之一(补充表2)。通过热稳定性分析,发现ISP I在一系列升高的温度下保护了SELH(图3B),这表明ISP I以SELH为靶点。为了验证ISP I靶向SELH的特异性,设计了一种表面等离子体共振(SPR)方法来评估ISP I与细菌中合成的过氧化物酶的相互作用,包括SELH、硫氧还蛋白(TrxA)、过氧化氢酶(KatA)和硫醇过氧化物酶(TpX)。ISP I与SELH紧密结合,但不与其他过氧化物酶结合(图3C)。ISP I在多种肿瘤细胞株中以剂量依赖的方式降低了SELH蛋白的表达(图3D和E)。环己酰亚胺(CHX)追踪实验证实,ISP I处理降低了SELH蛋白的半衰期,这表明ISP I促进了SELH蛋白的降解(图3F和G)。总的来说,这些结果表明,ISP I靶向SELH并促进SELH蛋白降解。
为了证实ISP I通过SELH依赖机制抑制细胞生长,用CRISPR-Cas9生成了SELH缺失的LN229和RCC细胞株(分别为786-O和RCC4)。与野生型细胞相比,SELH缺陷细胞能抵抗ISP I处理(图3H)。使用选择性siRNA敲除,SELH抑制显著降低了细胞生长速率,延缓了细胞增殖,增加了细胞凋亡(图3I-K)。为了评估SELH是否在体内调节肿瘤生长,将野生型或SELH缺失的LN229细胞原位注射给NSG小鼠:结果发现SELH-deficient小鼠总生存期增加了近两倍(SELH-deficient LN229组:野生型LN229组=51天:28天,图3L)。C57BL/6小鼠注射野生型B16细胞或SELH缺陷B16细胞(图3M - O)。尾静脉接种12天后,与注射野生型B16细胞的小鼠相比,SELH缺陷B16细胞的小鼠患肺肿瘤的几率显著降低(图3N-O)。因此,ISP I通过抑制SELH抑制原发和转移性肿瘤的生长。
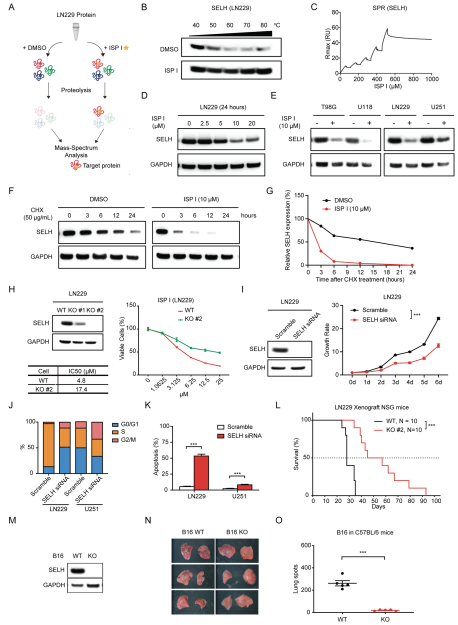
图3 ISP I以SELH为目标
4. ISP I破坏细胞内氧化还原稳态
由于硒蛋白可以保护机体免受活性氧的侵害,因此研究ISP I是否改变了体外氧化还原稳态。首先,评估ISP对谷胱甘肽过氧化物酶活性的影响:ISP I处理的LN229细胞谷胱甘肽过氧化物酶活性显著降低(图4A)。ROS-Glo H2O2检测证实,ISP处理过的肿瘤细胞内ROS水平升高(图4B)。MitoSOX染色,然后流式细胞仪检测,显示ISP I处理后的细胞内ROS升高,且呈剂量依赖性(图4C和D)。
下一步评估ISP I是否改变抗氧化下游信号通路。经ISP I处理的LN229细胞的转录组分析和GSEA均显示氧化途径的广泛改变以及核因子红系2相关因子2 (NFE2L2/NRF2)的特异性上调(图4E和F)。在ISP I处理的细胞中,NFE2L2的表达显著上调;ISP I诱导NFE2L2信号通路下游靶蛋白HMOX1和NQO1的增加(图4G和H)。过表达SELH降低了NFE2L2蛋白水平,而在SELH沉默的细胞中NFE2L2蛋白水平升高(图4I)。NFE2L2 mRNA在SELH沉默的U251细胞中略有增加(图4J)。SELH沉默的细胞HMOX1 mRNA显著增加,与NFE2L2蛋白的增加一致(图4K)。通过抑制SELH, ISP I诱导ROS积累并激活抗氧化信号通路,包括NFE2L2通路。
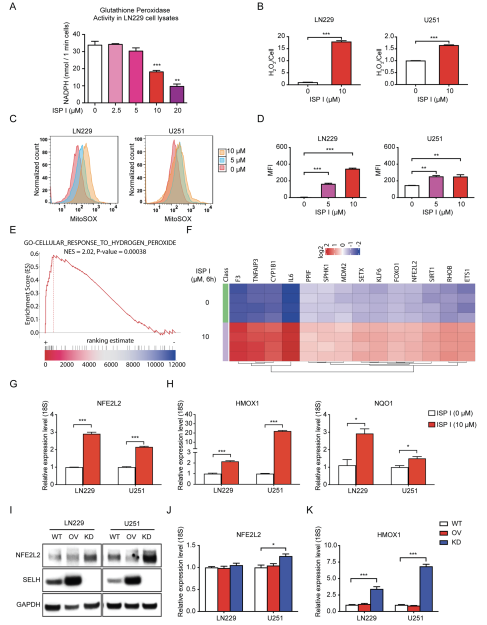
图4 ISP I触发ROS积累和氧化应激反应通路
5. ISP I触发DNA损伤和R-环的形成
由于细胞内ROS水平的增加导致氧化性DNA损伤,从而诱导基因组不稳定性并抑制细胞周期进展,因此评估ISP I治疗是否增强了癌细胞的DNA损伤。免疫荧光染色和western blot分析显示,ISP I处理的细胞显示出较高数量的γH2AX,这是DNA损伤的敏感标记(图5A和B)。ISP I处理的细胞以剂量依赖性的方式显示R-loop积累增加(图5C和D)。ISP I以剂量依赖性的方式减少EdU掺入DNA(图5C和I)。γH2AX和EdU显示,ISP I处理不仅持续增加DNA损伤和减少DNA复制,而且改变R-环定位(图5C, E-J)。在ISP I处理的U251细胞中,R-环病灶集中在核浆而不是核仁(图5C)。R-环位点特异性地定位于DNA损伤位点,而不是DNA复制位点(图5G和J)。此外,在ISP i处理的U251细胞中发现了核仁离域,这是核仁应激的证据(图5C)。 总的来说,ISP I显著增加细胞内ROS水平,降低DNA复制,导致DNA损伤和R-环的形成。
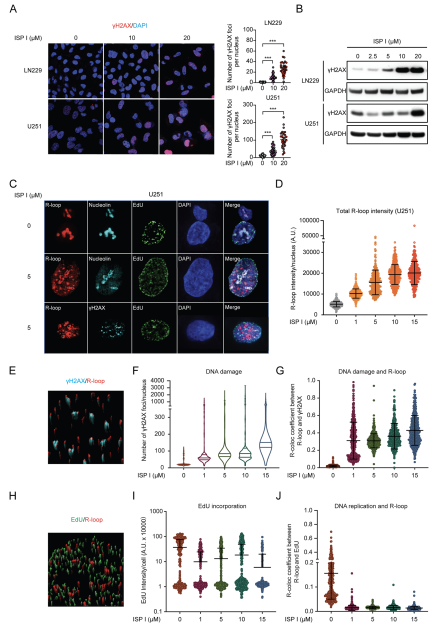
图5 ISP I触发DNA损伤和R环形成,并改变R环的定位
6. ISP I通过JNK2/TIF-IA通路抑制核仁rRNA转录
免疫荧光成像显示,ISP I处理的LN229细胞核仁蛋白分散度增加,核仁内SELH表达显著降低(图6A)。ISP I处理LN229细胞后,NPM1蛋白水平下降,p53(凋亡标志物)升高,且呈剂量依赖性(图6B)。GSEA分析证实了p53通路的诱导(图6C和D)。为了评估核糖体生物发生的改变并探索与核仁功能障碍的潜在联系,接着分析了pre-rRNA转录:ISP I以剂量依赖的方式减少pre-rRNA(POLI转录本)(图6E)。ISP I以剂量依赖的方式增加JNK2磷酸化,表明细胞ros诱导的JNK2激活升高(图6F)。虽然ISP I处理导致了POLI和TIF-IA表达的轻微下降趋势,但免疫共沉淀分析显示,在ISP I处理或SELH缺失的LN229细胞中,TIF-IA和POLI之间的生理相互作用被显著破坏(图6G):提示激活的JNK2在SELH缺失细胞中主要削弱了TIFI-IA和POLI的相互作用。CHIP分析显示,在ISP I处理或SELH缺陷的LN229细胞中,POLI补充到启动子和rDNA编码区的量减少(图6H)。这表明,ISP I通过激活ROS/JNK2/TIF-IA通路,抑制了SELH受损的POLI-rDNA相互作用。这些发现表明,ISP I通过抑制POLI转录而破坏了核糖体的生物发生,但并不改变成熟的RNA机制。

图6 ISP I诱导核仁应激反应
结论:
一种新型的重组大环内酯类抗生素通过抑制特定蛋白质SELH在核仁中发挥有效的抗癌作用。这会破坏RNA聚合酶I,导致轻微但灾难性的DNA损伤,并导致细胞周期停滞和肿瘤细胞凋亡,从而造成癌细胞的脆弱性。ISP I对小鼠正常组织的有害影响最小,专门针对SELH。通过利用SELH抑制作用,ISP I揭示了肿瘤细胞中一种独特的脆弱性。通过利用SELH抑制,将ROS稳态的紊乱与核糖体合成功能障碍联系起来,ISP I揭示了肿瘤细胞特有的脆弱性。因此,核仁和SELH是有希望的癌症治疗靶点。
参考文献:
Cui J, Zhou J, He W, et al. Targeting selenoprotein H in the nucleolus suppresses tumors and metastases by Isovalerylspiramycin I. J Exp Clin Cancer Res. 2022; 41(1):126. doi: 10.1186/s13046-022-02350-0.