






EZH2通过整合素β1-FAK激活TGFβ信号促进乳腺癌骨转移
乳腺癌是全世界女性中最常见的癌症。大约50-70%的乳腺癌晚期患者发生骨转移,导致骨骼相关事件,包括疼痛、病病性骨折、脊髓受压、高钙血症和其他并发症。在本研究中,作者探讨了骨转移的分子机制,可能为骨转移患者提供额外的治疗策略。本文于2022年5月发表于《Nature communications》, IF=12.121。
本文技术路线:

本文主要内容:
1. EZH2促进乳腺癌骨转移,EZH2甲基转移酶抑制剂不能阻止这一过程
为了探讨EZH2在乳腺癌骨转移中的作用,作者构建了EZH2敲除细胞系1566.shEZH2及其对照细胞系1566.shScr。与注射1566.shScr细胞的小鼠相比,注射1566.shEZH2的小鼠无骨转移生存期和总生存期更长(Fig. 1a)。 生物发光成像(BLI)、x射线成像和苏木精和伊红(H&E)染色均显示出相同的结果(Fig. 1b)。
使用CRISPR/CAS9系统,生成了ezh2敲除的MDA-MB-231细胞亚克隆(231.KO)及其对照克隆 (231.sgCtrl)。其中的231.KO#1与野生型EZH2(231.KO# 1.EZH2) 或pLenti对照载体(231.KO#1.pLenti)中稳定表达。231.KO# 1.EZH和231.KO#1.pLenti)分别注射到小鼠中,注射231.KO#1.EZH2的细胞用载体或GSK126处理,GSK126是一种有效的EZH2小分子甲基转移酶抑制剂,而对照组小鼠注射231.KO#1.pLenti治疗。结果表明,231.KO#1.EZH2组无骨转移生存率明显低于231.KO#1.pLenti组(Fig. 1c, d)。出乎意料的是,gsk126处理的231.KO#1.EZH2组无转移生存率与vehicle处理的231.KO#1.EZH2 组相似(Fig. 1c, d)。与对照组相比,注射1566.KO细胞的小鼠无转移生存期和总生存期均显著延长 (Fig. 1e,f)。对照细胞组小鼠在无骨转移生存期、总生存期方面,与EPZ-6438组之间没有显著差异(Fig. 1e, f)。在体外实验中,高表达ezh2的细胞比低表达ezh2的细胞具有更强的迁移和侵袭能力(Fig. 1g)。与对照231.KO#1相比,野生型EZH2的重表达(231.KO#1. ezh2)显著提高了细胞迁移和侵袭能力。此外,与注射231.KO#1.pLenti细胞的对照组相比,野生型EZH2的重表达(231.KO#1.EZH2)显著增加细胞迁移和侵袭(Fig. 1h)。
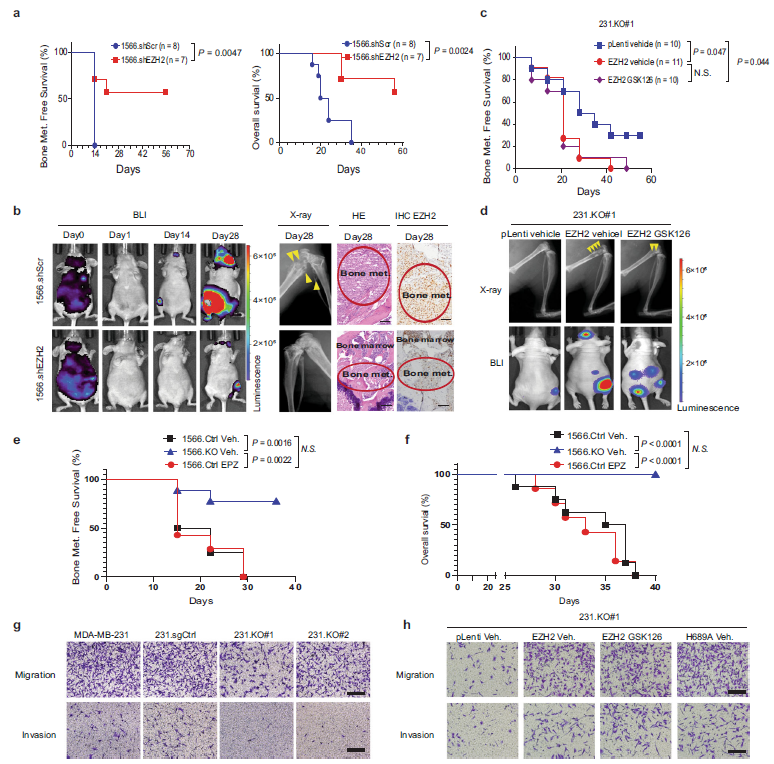
Fig1 EZH2促进乳腺癌骨转移,EZH2甲基转移酶抑制剂不能阻止这一过程
2. EZH2调节乳腺癌骨转移的恶性循环
乳腺癌细胞与RAW264.7破骨前体细胞和MC3T3成骨细胞共培养(Fig. 2a)。共培养六天,与MDA-MB-231 和 231.sg对照细胞相比,sezh2基因敲除的231.KO # 1和231.KO#2细胞的生长明显受到抑制(Fig. 2b)。与EZH2敲除的细胞共培养时,与MDA-MB-231 和 231.sg对照细胞相比,成熟破骨细胞的前体破骨细胞RAW264.7明显减少(Fig. 2c)。与231.KO#1相比,231. ko # 1.H689A细胞重新表达H689A促进了肿瘤细胞增殖和破骨细胞成熟(Fig. 2d, e)。静脉注射231.KO#1.EZH2和231. ko # 1.H689A细胞转染小鼠,观察骨转移生长情况,发现231.KO#1.EZH2和231. KO # 1.H689A细胞诱导骨转移病变(Fig. 2f)。PTHLH是乳腺癌骨转移的介质。通过qRTPCR检测,在MDA-MB-231和4T1细胞中敲除EZH2可以降低TGFβ处理下的PTHLH mRNA表达, 而GSK126处理4T1细胞并没有降低Pthlh mRNA的表达(Fig. 2g, h).
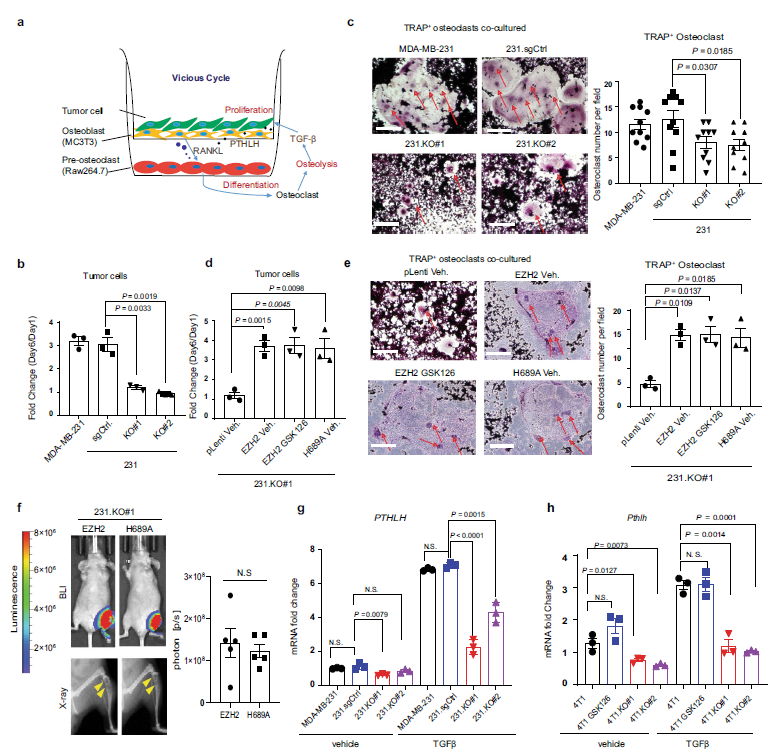
Fig2 EZH2调节乳腺癌骨转移的恶性循环
3. FSP1EZH2在TGFβ刺激下增加pS465/467-Smad2和pY397-FAK水平
PTHLH是一个众所周知的TGFβ下游基因,受p-Smad2/Gli2转录因子复合体或p38 mapk7,23调控。为了进一步探索EZH2是如何促进乳腺癌细胞中PTHLH和TGFβ信号转导的,作者检测了乳腺癌细胞中pS465/467-Smad2和pT180/Y182-p38 MAPK水平。在TGFβ刺激下,MDA-MB-231细胞敲除EZH2可抑制pS465/467-Smad2水平,但Smad2、Smad3、Smad4蛋白水平没有显著变化(Fig. 3a)。与对照载体相比,H689A EZH2重表达也增加了pS465/467-Smad2的水平(Fig. 3b)。敲除ezh2基因后进行反向蛋白芯片分析,结果显示,显著下调和上调的蛋白为激酶,包括酪氨酸激酶(Fig. 3c)。值得注意的是,FAK上酪氨酸397的磷酸化水平(pY397-FAK)显著降低(Fig. 3c)。通过WB验证了RPPA数据,结果显示,在MDA-MB-231和MCF7细胞中敲除EZH2可以抑制TGFβ处理下的pY397-FAK和pS465/467-Smad2水平(Fig. 3d)。
为了检查pY397-FAK的增加是否与pS465/467-Smad2的增强有关,作者用不同浓度(1-10 μM)的FAK抑制剂(FAKi、VS-4718或VS-6063),再经TGFβ处理(5 ng/ mL, 2 h) MDA-MB-231细胞,检测pS465/467-Smad2。结果发现FAKi降低了pS465/467-Smad2水平,甚至在TGFβ刺激下也降低了PTHLH mRNA的表达 (Fig. 3e)。此外,在MDA-MB-231细胞中使用shRNAs敲除FAK (shFAK#2或shFAK#3)也能抑制pS465/467-Smad2的水平(Fig. 3f)。
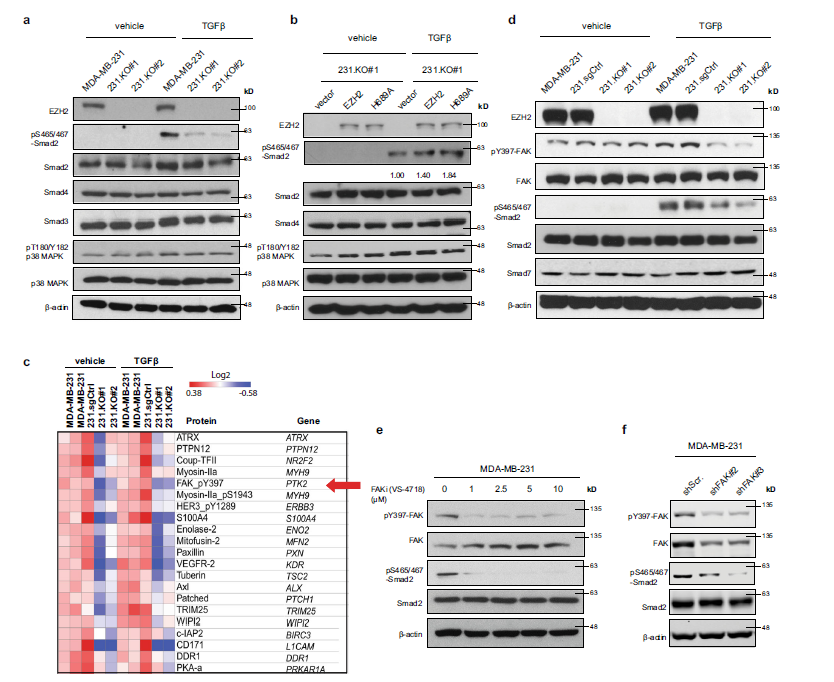
Fig3 EZH2在TGFβ刺激下增加pS465/467-Smad2和pY397-FAK水平
4. pY397-FAK诱导TGFβ ri酪氨酸磷酸化,增强其与TGFβ rii的结合,以响应TGFβ
在mda - mb - 231细胞中,评估FAK是否调控TGFβRI和TGFβRII的表达。发现无论有无TGFβ暴露,FAK IP均可降低TGFβRI,而非TGFβ RII (Fig. 4a)。此外,通过FAKi VS-4718处理阻断FAK激酶活性,降低了FAK与TGFβRI的结合(Fig. 4b)。TGFβRII的WB结果显示FAK抑制显著降低了TGFβRI与TGFβ RII的结合,以响应TGFβ的刺激(Fig. 4c)。此外,作者检测到TGFβRI上的酪氨酸磷酸化, FAKi VS-6063处理降低了酪氨酸磷酸化(Fig. 4d)。接下来,作者发现了一个未报道的酪氨酸182 (pY182)上的TGFβRI磷酸化位点,位于甘氨酸和丝氨酸残基富集区域(Fig. 4e)。
结构分析显示,TGFβRI的Y182位点高度暴露于潜在的磷酸化(Fig. 4f),靠近TGFβRI的两个苏氨酸位点和一个丝氨酸位点(T185、T186、S187) (Fig. 4g),它们被TGFβRII31和32结合并磷酸化。蛋白质对接结果表明,TGFβRI的Y182指向TGFβRII的K381(Fig 4g)。

Fig 4 pY397-FAK诱导TGFβ ri酪氨酸磷酸化,增强其与TGFβ rii的结合,以响应TGFβ
5. EZH2增加FAK上游ITGB1的表达
作者发现,敲除EZH2导致RNA Pol II与ITGB1(编码整合素β1)启动子的结合显著降低,而与ITGB3(编码整合素β3)的结合变化不大(Fig. 5a)。qRT-PCR显示敲低或敲除EZH2下调ITGB1 mRNA表达(Fig. 5b, c)。同时,ITGB1启动子驱动的荧光素酶报告基因检测显示,与与231.KO#1 和 231.KO#2相比,ezh2表达的MDA-MB-231 和 231.sgCtrl 细胞ITGB1启动子活性较高(Fig. 5d)。
ChIP-qPCR中,使用一系列的PCR引物结合到ITGB1启动子的不同区域,结果发现在MDA-MB-231细胞中,EZH2被招募到ITGB1启动子从P1到P5位点,预期在231.EZH2与ITGB1启动子位点的结合缺失(Fig. 5e)。在MDA-MB-231细胞中。EZH2敲除后,RNA Pol II与ITGB1启动子的结合也失去了(Fig. 5f)。类似地,在231.shEZH2#3 和231.shEZH2#4细胞中敲低EZH2能降低ITGB1启动子P1到P4位点的RNA Pol II结合(Fig. 5g)。
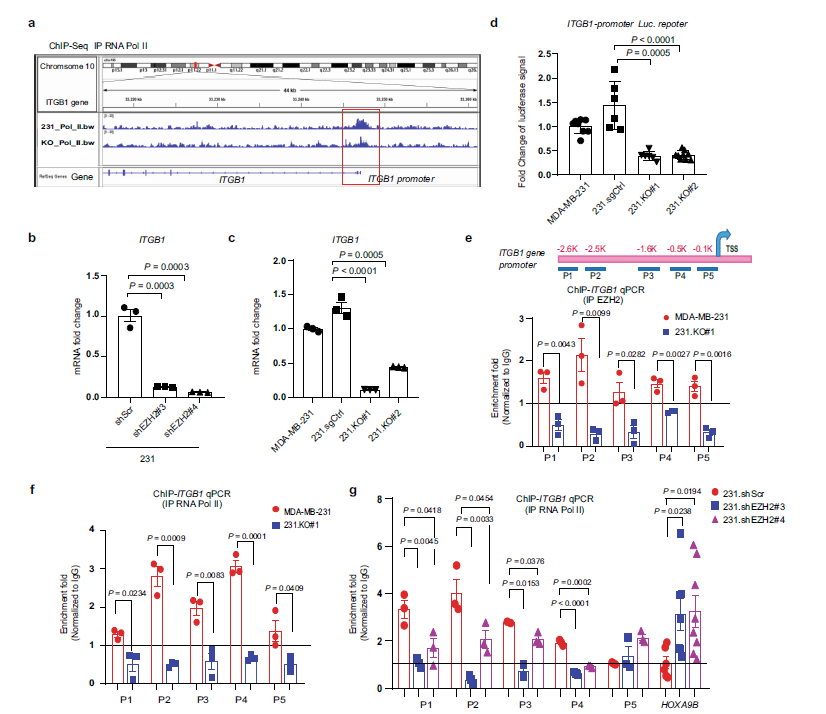
Fig5 EZH2增加FAK上游ITGB1表达
6. 临床适用的FAK抑制剂能阻断治疗ezh2诱导乳腺癌骨转移
使用BLI检测产生的骨转移生长,这证实了GSK126治疗没有阻止骨中的肿瘤生长. 令人兴奋的是,与对照组相比,FAKi VS-6063治疗显著阻碍了骨肿瘤的生长(P = 0.0442)(Fig 6a)。骨转移免疫组化染色显示FAKi VS-6063显著降低pS465/467-Smad2水平(P = 0.0066)和骨转移灶中PTHLH表达(P = 0.0074),两种药物均有效抑制其靶点(Fig. 6b, c)。最后,作者检查了GSE2603数据集并验证了EZH2表达与乳腺癌患者无骨转移生存期呈负相关(r =−0.2394,P = 0.03) (Fig. 6d), 提示EZH2在原发性乳腺肿瘤中的高表达会导致患者发生骨转移的高风险。作者还研究了EZH2表达与下游效应因子的相关性,GSE14020数据集中乳腺癌患者骨转移组织中PTHLH的含量。作者发现骨转移患者中EZH2 mRNA表达与PTHLH mRNA表达呈正相关(Fig. 6e)
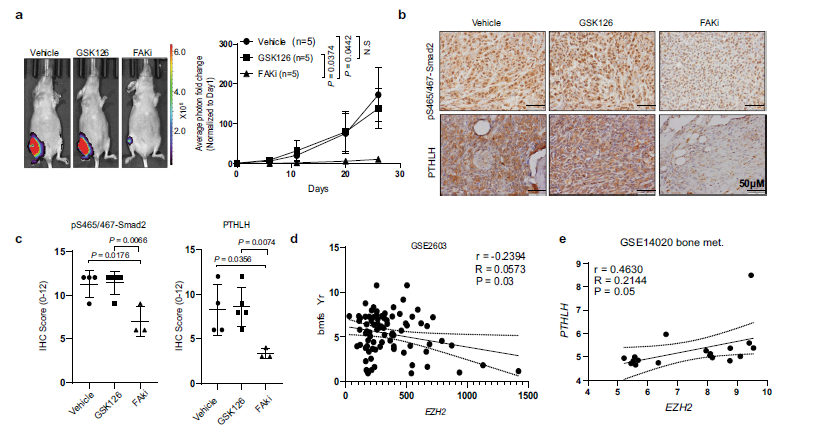
Fig6临床适用的FAK抑制剂治疗阻断ezh2诱导的乳腺癌骨转移
综上所述,作者发现FAK是EZH2在乳腺癌骨转移恶性循环中的下游效应因子。发现FAKi联合标准的抗骨吸收药物、化疗或放疗可能为骨转移的乳腺癌患者提供额外的好处。