






FTO通过m6A-YTHDF2依赖性诱导PDGFC自分泌活性促进胰腺癌进展
m6A是一种新兴的mRNA修饰调节因子,在肿瘤发生中发挥着新的作用。尽管m6A修饰在病理和生理过程中都具有重要的功能意义,但其在胰腺导管癌(PDAC)中的作用仍不明确。在这里,我们发现FTO表达与PDAC患者的不良预后相关,并且抑制FTO表达抑制细胞增殖。此外,FTO直接靶向PDGFC,并以m6A-YTHDF2依赖的方式稳定其mRNA表达。PDGFC上调导致Akt信号通路重新激活,促进细胞生长。总之,我们的研究表明,FTO下调导致PDGFC 3ʹUTR中m6A修饰增加,然后以m6A-YTHDF2依赖性方式调节其转录水平的降解,突出了PDAC治疗和预后预测的潜在治疗靶点。本文于2022年4月发表于“Oncogene”(IF=9.867)上。
技术路线

结果
1)FTO在PDAC中高表达,与不良预后相关
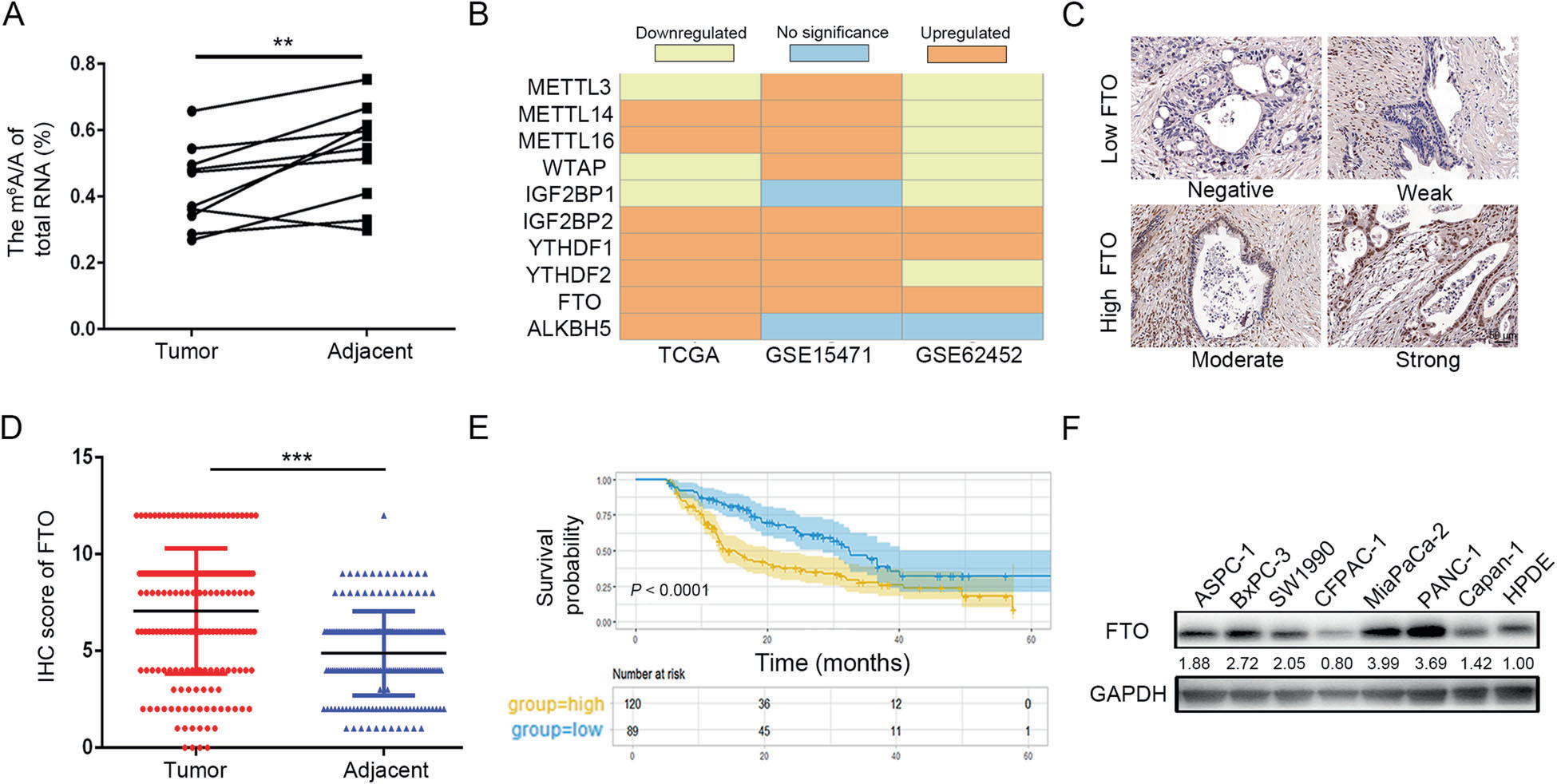
为了解释m6A在PDAC中的关键作用,我们通过m6A比色分析检测了10例新鲜人胰腺肿瘤组织及其相应正常组织中m6A的整体水平。初步观察发现,与相应的正常组织相比,PDAC组织中整体m6A丰度显著降低(图1A)。然后,为了评估PDAC中主要m6A修饰酶的表达谱,我们分析了TCGA、GTEx、GSE15471和GSE62452的临床数据集,发现PDAC组织中FTO mRNA表达水平显著高于正常组织(图1B)。此外,我们验证了复旦大学上海癌症中心(FUSCC)队列中的生物信息学数据。对含有209名患者样本的组织微阵列(TMA)进行免疫组织化学(IHC)染色(图1C)。根据IHC评分,与正常组织相比,FTO在肿瘤组织中的表达水平更高(图1D)。Kaplan–Meier生存曲线显示,FTO高表达与PDAC患者预后不良相关(图1E)。最后,我们发现,与人胰腺导管上皮(HPDE)细胞相比,多个PDAC细胞系中FTO蛋白表达上调(图1F)。
2)沉默FTO抑制PDAC肿瘤生长
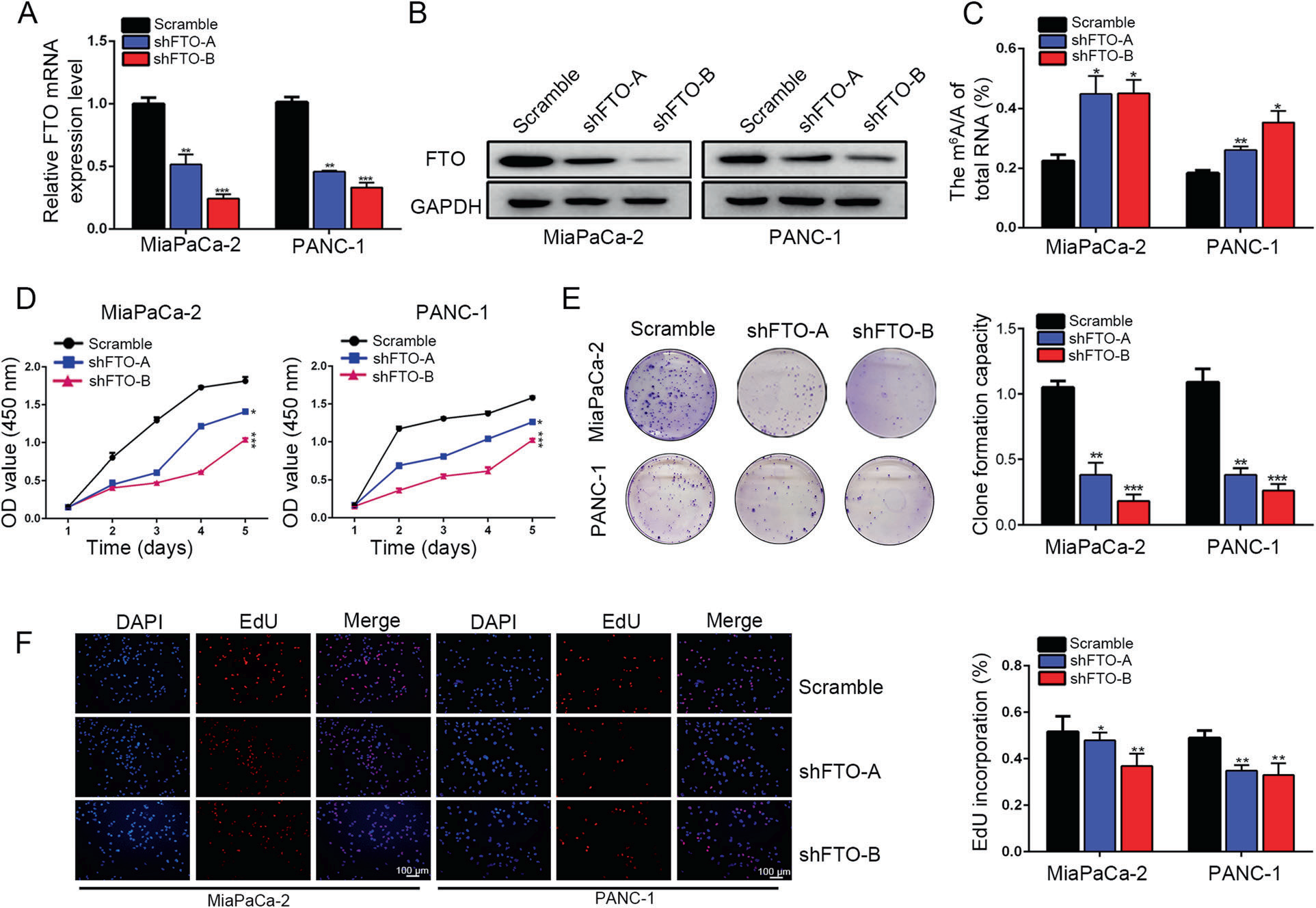
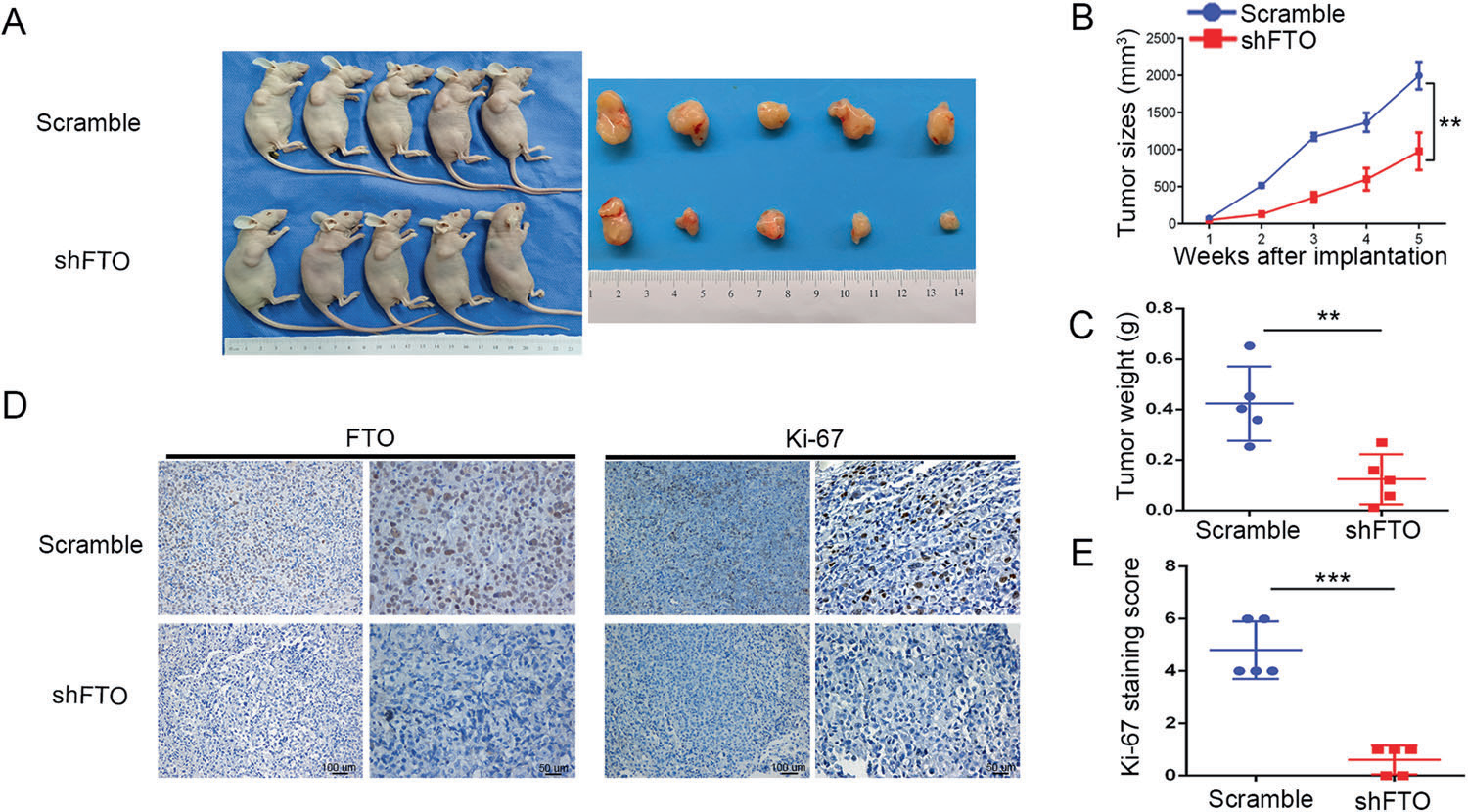
为了研究FTO在PDAC中的作用,用shFTO-a和shFTO-B转染PANC-1和MiaPaCa-2细胞。FTO敲除效应在mRNA和蛋白质表达水平均得到验证(图2A,B)。FTO敲除导致m6A水平增加(图2C)。FTO敲除显著降低PDAC细胞增殖(图2D),并降低集落形成效率(图2E)。同样,EdU染色分析表明,FTO敲除大大降低了EdU阳性细胞的百分比(图2F)。接下来,我们植入了一个皮下胰腺异种移植瘤,以进一步确定FTO在体内的致癌作用。将具有稳定FTO敲除(shFTO-B)的MiaPaCa-2细胞皮下注射到4周龄雌性BALB/c裸鼠体内(图3A)。与体外观察结果一致,FTO沉默组的肿瘤生长大小和重量明显低于对照组(图3B,C)。随后使用抗FTO抗体和增殖标记物Ki-67进行IHC染色,结果表明FTO敲除显著降低了shFTO组的Ki-67染色(图3D,E)。因此,FTO在促进PDAC肿瘤生长中起着关键作用。
3)通过m6A-Seq识别FTO下游靶点
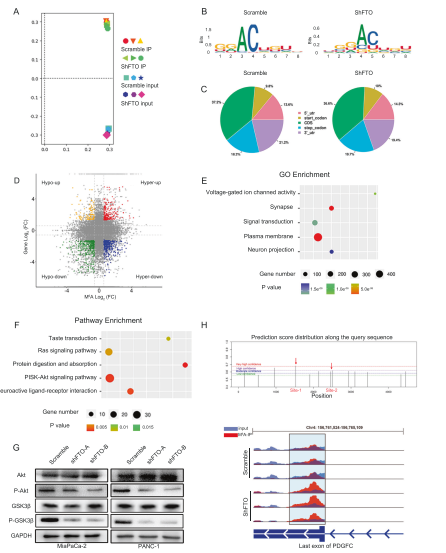
为了确定PDAC中的FTO下游靶点,我们进行了甲基化RNA免疫沉淀测序(MeRIP-seq)。主成分分析(PCA)显示,每个样本有3个重复聚类在一起,表明两组之间重现性良好(图4A)。在对照和FTO敲除细胞中,GGAC基序在m6A中高度富集(图4B)。根据m6A-seq的结果,我们进一步发现m6A峰主要富集在相邻的编码序列(CDS)和3’UTR(图4C)。我们进一步研究了MeRIP-seq表达数据,并探索了m6A和RNA水平均发生显著变化的峰值比例。事实上,在两组中检测到292个具有高甲基化m6A峰值以及mRNA表达降低的基因(图4D)。在这些差异表达基因(DEG)中,电压门控离子通道活性、信号转导和质膜是GO富集分析中最丰富的条目(图4E)。KEGG富集分析表明,PDAC中PI3K/Akt通路与FTO依赖转录相关(图4F)。为了验证这一发现,我们检测了Akt、P-Akt和P-GSK3β的表达(图4G)。FTO敲除显著降低了PANC-1和MiaPaCa-2细胞中这些基因的磷酸化水平。在MiaPaCa-2和PANC-1细胞中,scramble和FTO基因敲除组的PDGFC表达差异最大。最重要和有趣的是,在FTO敲低组中,一些下调的转录本表现出高甲基化的m6A修饰,包括PDGFC。我们关注PDGFC m6A峰,并发现其mRNA的3ʹUTR周围的富集在FTO敲除后增加(图4H)。因此,PDGFC被选为FTO介导的m6A 修饰的候选靶点。
4)FTO缺失以m6A-YTHDF2依赖的方式下调PDGFC mRNA水平
MiaPaCa-2和PANC-1细胞中的FTO敲除降低了PDGFC mRNA和蛋白质水平(图5A,B)。通过qRT-PCR(图5C),在异种移植物中也证实了类似的趋势。此外,与IgG RIP组相比,FTO敲除显著促进PDGFC mRNA中的m6A水平(图5D)。与对照细胞相比,FTO敲除细胞的荧光素酶活性显著降低。PDGFC-MUT2几乎消除了这种诱导,表明PDGFC表达的调节受FTO相关m6A修饰的控制(图5F)。由于YTHDF2作为主要m6A读取器的特异性,我们假设YTHDF2在PDGFC中的RNA识别和相互作用水平发挥作用。通过评估TCGA和GEO数据库,我们发现与正常组织相比,PDAC组织中的YTHDF2上调。事实上,通过RNA免疫沉淀(RIP)分析评估,YTHDF2与PDGFC有强烈的相互作用。当FTO被敲除时,PDGFC转录本中的YTHDF2富集显著减少(图5G)。此外,我们在FTO减少的MiaPaCa-2细胞中敲除了YTHDF2,并观察到胰腺细胞中PDGFC表达的部分恢复(图5H,I)。在FTO敲除条件下,YTHDF2敲除也消除了PDGFC mRNA减少的稳定性(图5J)。因此,我们的数据表明FTO以m6A-YTHDF2依赖的方式调节甲基化PDGFC mRNA水平。

5)PDGFC是一种促癌因子,与PDAC患者的FTO表达呈正相关
TCGA数据分析表明,PDGFC水平与PDAC患者的OS呈负相关,PDGFC和FTO水平之间呈显著正相关(图6A,B)。值得注意的是,较高的PDGFC水平与较差的OS显著相关(图6C),并与FTO表达呈正相关(图6D)。此外,利用在TMAs上进行的IHC染色,我们进一步对PDGFC进行存活分析,并验证其在肿瘤和相邻正常组织之间的表达差异(图6F,H)。此外,如图6G所示,我们还生成了一个包含PDGFC和FTO的新IHC面板来预测PDAC的预后。Kaplan–Meier生存分析表明,两种标记物高表达的患者OS最短。
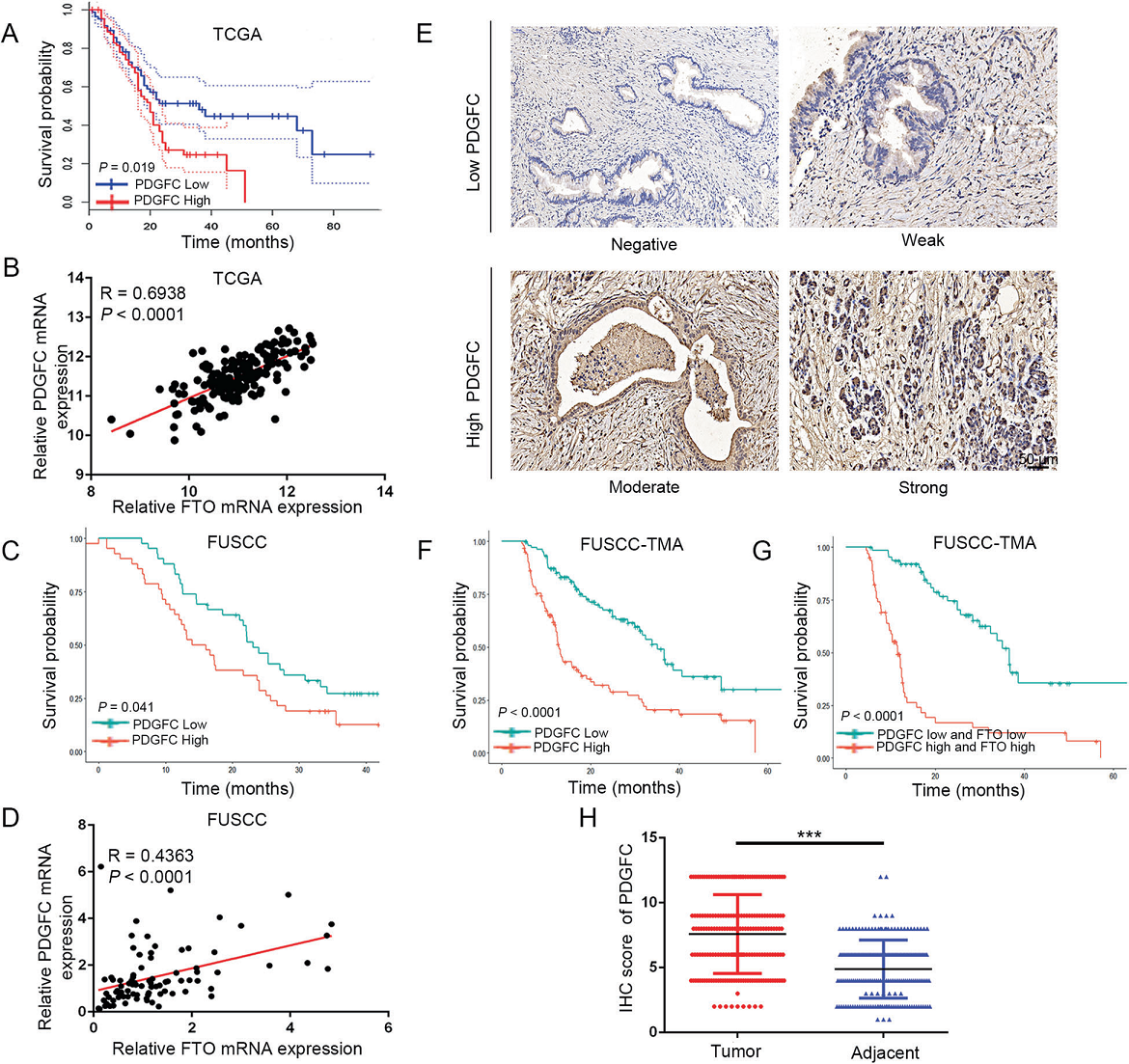
6)FTO介导的PDGFC激活与PDAC的肿瘤发生有关
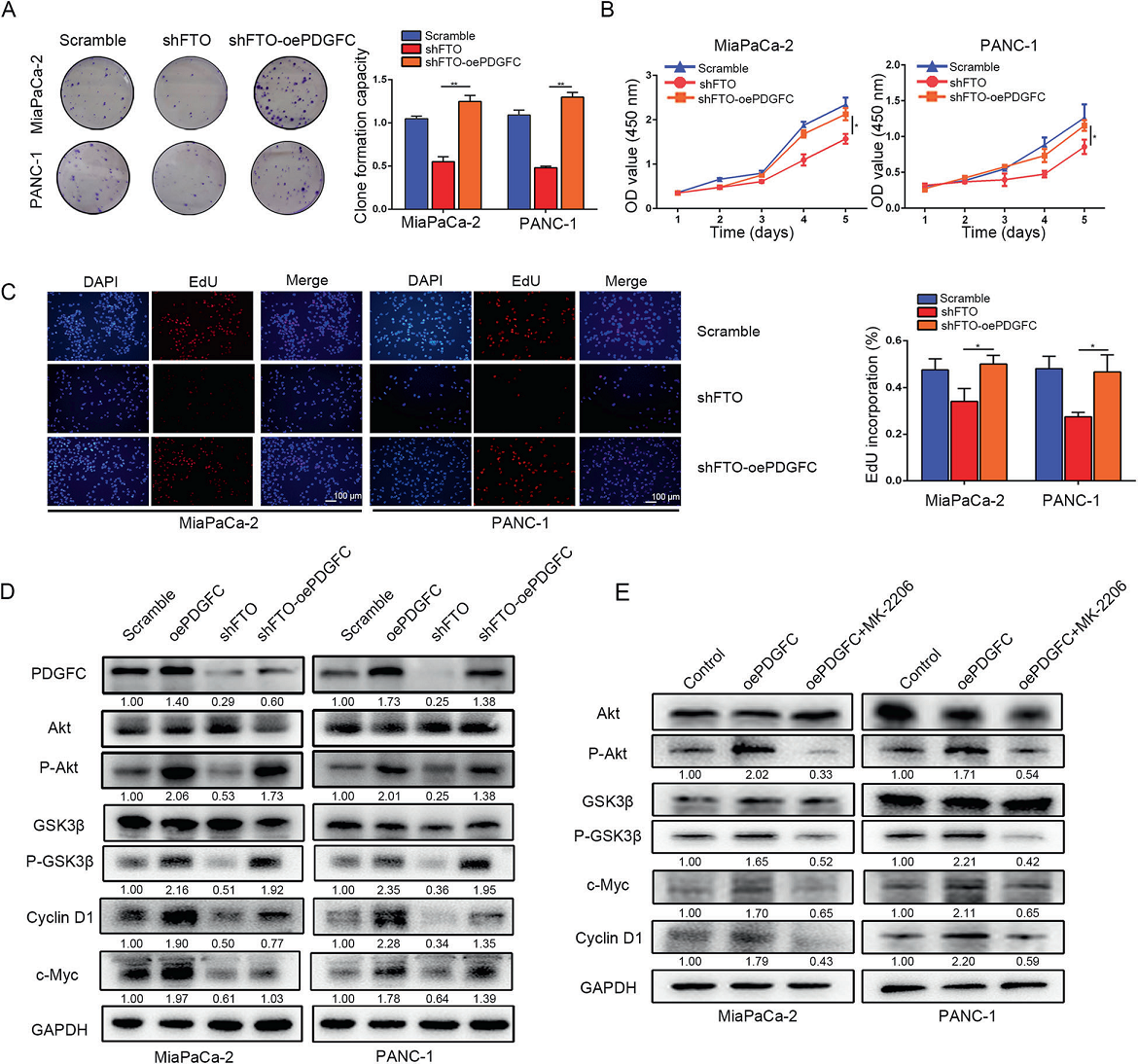
为了确定PDGFC是否介导FTO在PDAC生长和进展中的作用,我们首先基于PDGFC的内源性表达将编码人PDGFC插入物的慢病毒载体转染到细胞中。我们发现PDGFC的过表达特别增加了PDAC细胞活力和集落形成,但也消除了FTO敲除条件下的下降(图7A-C)。接着,我们评估了胰腺癌细胞中Akt/GSK3β磷酸化、c-Myc和Cyclin D1的表达。FTO敲除降低了这些基因的表达,而PDGFC过表达挽救了这种趋势(图7D)。此外,为了进一步证实我们的假设,我们检测了Akt抑制剂(MK2206)是否可以消除PDGFC过表达细胞中这些基因的表达。在PDGFC过表达的细胞中,Akt/GSK3β磷酸化水平以及c-Myc和Cyclin D1表达上调,而在MK2206处理的细胞中,这些水平降低(图7E)。
7)FTO诱导PDGFC自分泌活性并与PDAC细胞增殖相关
我们在FTO稳定的敲除细胞中过表达PDGFC,并使用ELISA检测PDAC细胞分泌PDGFC的内源性水平(图8A)。正如预期的那样,稳定敲除FTO后PDAC细胞释放的PDGFC减少,过表达后PDGFC恢复。FB23-2是一种直接与FTO结合并选择性抑制FTO m6A去甲基酶活性的抑制剂,它确实抑制PDAC细胞中PDGFC的分泌(图8B)。CCK-8分析表明,CFPAC-1和MiaPaCa2细胞中FTO的过表达增强了细胞的增殖和活力,而FTO的过表达与PDGFR抑制剂的结合显示出更强的抑制作用(图8C,D)。此外,对在DMEM(全培养基)中培养的FTO scramble和敲除细胞进行CCK8分析,无论是否用重组人PDGFC(10 ng /ml)处理,也证明了外源性PDGFC的刺激增殖活性(图8E)。为了进一步阐明PDGFC诱导的信号通路,我们用重组人PDGFC(10 ng/ml)处理MiaPaCa-2细胞。值得注意的是,48小时的重组人PDGFC预处理可在scramble和FTO敲除条件下诱导Akt/GSK3β激活的磷酸化水平(图8F)。总之,这些结果表明,自分泌内源性和外源性重组人PDGFC可促进PDAC细胞的生长,其部分由Akt/GSK3β信号驱动。
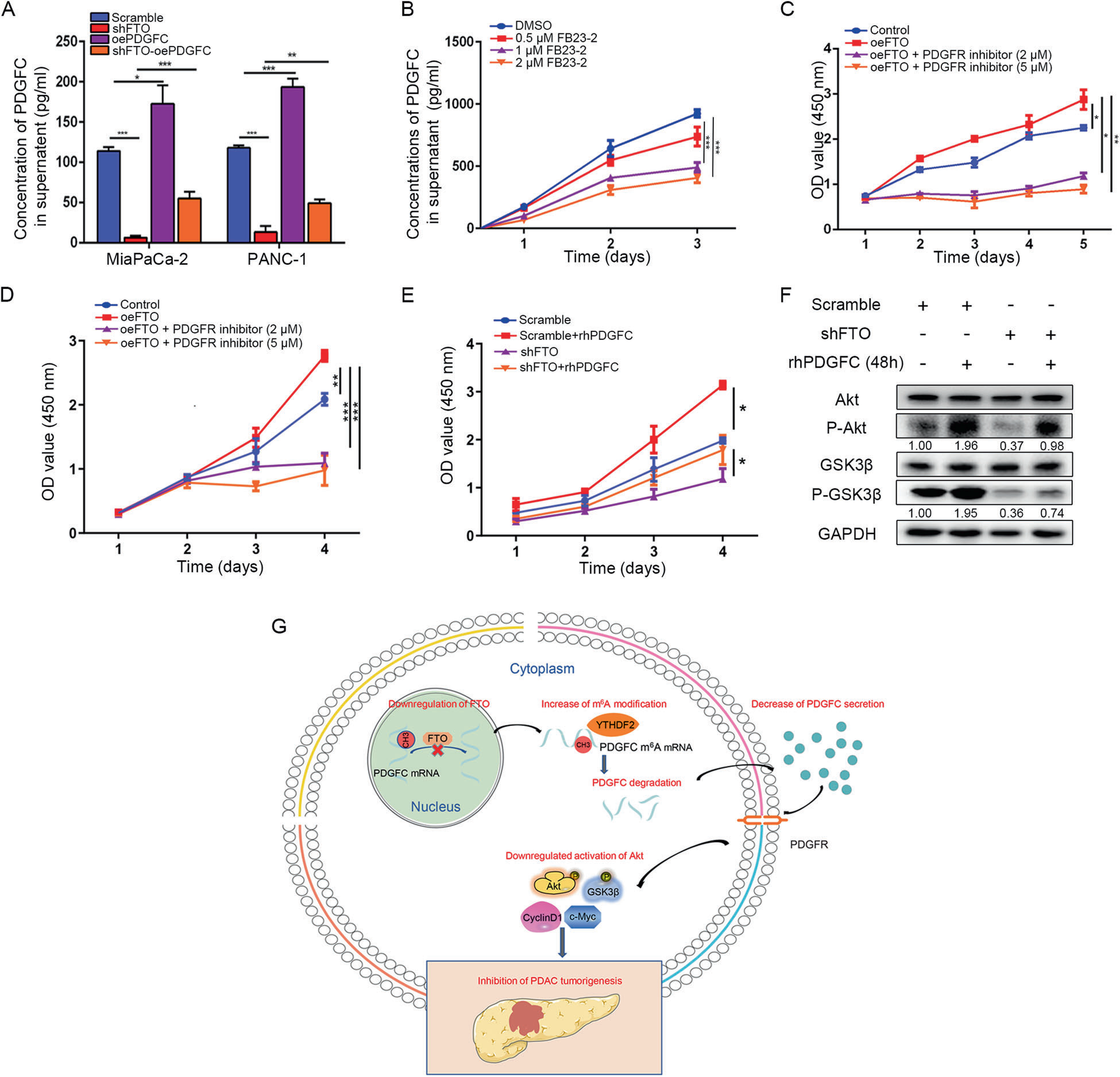
结论:我们的研究揭示了胰腺癌中m6A甲基化水平的降低是由FTO的失调引起的。FTO和PDGFC的联合可作为预后工具和治疗反应标志物。FTO/PDGFC/PDGFR治疗靶点的潜力可用于开发有效治疗PDAC的治疗方案。这些发现为m6A修饰调控PDAC肿瘤发生的分子机制提供了新的见解。
参考文献:
Tan Z, Shi S, Xu J, Liu X, Lei Y, Zhang B, Hua J, Meng Q, Wang W, Yu X, Liang C. RNA N6-methyladenosine demethylase FTO promotes pancreatic cancer progression by inducing the autocrine activity of PDGFC in an m6A-YTHDF2-dependent manner. Oncogene. 2022 May;41(20):2860-2872. doi: 10.1038/s41388-022-02306-w.