






17分生信——circRNA单细胞图谱首发
以往的研究表明,circRNAs在不同的组织和生物中具有高度特异性的表达,但circRNAs的细胞水平结构尚未完全确定。本文表征了circRNAs在人类和小鼠组织单细胞水平的表达图谱,将我们对circRNAs表达的了解扩展到单细胞水平,并构建了circRNAs的单细胞数据集的在线网站,为以这种前所未有的分辨率探索circRNAs提供了有用的资源。本文于2022年6月发表在《Nature Communications》IF:17.694期刊上。
技术路线

主要研究结果:
1、大规模单细胞研究显示circRNA具有高度细胞特异性
为了阐明circRNA的细胞构架,作者收集了171项涉及58种不同人类和小鼠组织或细胞类型的公开全长scRNA-seq数据集(图1a)。考虑到大多数3’RNA测序方法无法检测到缺乏poly(A)尾的circRNA,所以作者只收集全长测序技术的研究,然后使用嵌入多个最先进工具的综合管道计算基因和 circRNA 的单细胞水平表达值(图1b)。总之,40,604个人类和131,533个小鼠单细胞通过质量控制,并检测这些细胞中的circRNA进行下游分析。为了评估circRNA检测的可靠性,将所有的单细胞数据中的circRNA比对至circAtlas v2.0或其它数据库中。如图1c所示,在scRNA-seq队列中共检测到354,390个circRNA,其中76,824(21.67%)个circRNA可以在所有三个circRNA组中同时检测到。总之,32.43%的circRNA存在于这些批量RNA-seq数据库中,而其余67.57%的circRNA只在单细胞数据中检测到。值得注意的是,在circAtlas中唯一检测到的circRNAs比在circAtlas和单细胞数据集中共享的表达水平更低且长度更短(图1d,e)。这表明scRNA-seq可以有效捕获大多数高丰度circRNA。此外,通过MCS评分,这些共享的circRNA显示出很高的组织特异性,48.9%的共享的circRNA在两个以上物种中保守(MCS评分≥2),表明鉴定的circRNA具有很高的可靠性(图1f)。
对于所有在scRNA-seq数据集中检测到的circRNAs,其表达细胞数量与其平均表达水平之间正相关,一些高表circRNA如mmu-Cdr1_0001、mmu-Tulp4_0006和hsa-RIMS1_0021也在之前的研究中被报道(图1g)。再次证实了数据分析的可靠性。同时,在 scRNA-seq 数据中唯一检测到的 circRNA 通常在较少数量的细胞中表达(图 1h),但与其他数据库验证的 circRNA 相比具有相似的表达水平(图1i),提示这些circRNA具有高度的细胞特异性。特别是在人类和小鼠样本中,约 90% 的 scRNA-seq 特异性 circRNA 在不到 10 个细胞中表达,这使得使用bulk RNA-seq 技术几乎无法检测到(图1j)。综上所述,这些结果表明全长 scRNA-seq在揭示具有高细胞特异性的 circRNA 方面具有高灵敏度和可靠性,而由于在传统bulk RNA-seq 样本中表达细胞的比例相对较低,其中大部分可能被错误地忽略。此外,这些 scRNA-seq 特异性 circRNA 还在具有超过 10 个反向剪接读数的细胞中广泛表达(图1k)。
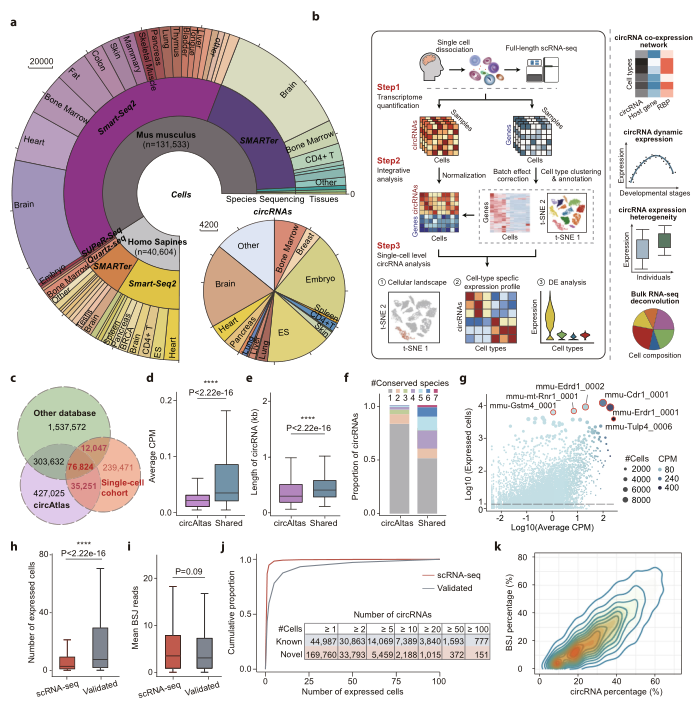
图1从单细胞测序数据集这发现circRNAs
2、脑circRNA在抑制性和兴奋性神经元中显示细胞特异性表达模式
为了研究 circRNA 的细胞景观,首先收集并分析了 18 项对小鼠大脑样本的研究,这也是收集的数据集中最大的队列,并分析和整合人类的脑细胞。共将41,911个细胞分为14个簇,检测到64,311个circRNA(图2a)。如图2b所示,大多数细胞聚集成GABA能神经元(GABA)、谷氨酸能神经元(GLUT)和小胶质细胞(MG)。 尽管这些簇中的细胞数量相似,但 GABA 能神经元和谷氨酸能神经元中circRNA尤其地丰富。作者对12个细胞特异性circRNA进行了PCR验证,并采用广泛使用的Tau方法检测了circRNA的细胞特异性,并将基因分为circRNA宿主基因和其他基因进行进一步的比较,如图2c所示,circRNA的特异性明显高于两组基因。同时,circRNA宿主基因的特异性也显着低于其他非宿主基因,因为circRNA往往来源于具有较高表达水平的基因,这导致细胞特异性相对较低。例如,在神经元细胞中特异性检测到来自小鼠 Taf1 基因的 12 个 circRNA 中的 10 个,并且在 GABA 能和谷氨酸能神经元中也观察到了不同的表达模式(图 2d)。
为了进一步验证circRNA在人脑中的表达谱,收集4个人脑scRNA-seq数据集,如图 2e 所示,具有较高表达水平的 circRNA 在两个物种中更可能是保守的,而物种特异性 circRNA往往具有较低的表达水平。与之前的结果一致,这些保守的circRNA中的大多数在 GABA 能和谷氨酸能神经元中高度富集,并且一部分circRNA也表现出在所有类型的细胞中普遍表达(图 2f)。circRNA的表达水平与RNA结合蛋白(RBP)的活性密切相关,作者计算了所有circRNA与所有细胞中circRNA宿主基因或RBP之间的Spearman相关系数并进行比较,结果发现circRNA与RBP之间的相关性显著高于宿主基因(图2g),尤其是PTBP1和PTBP2和circRNA高度相关。如预期的,在大多数细胞类型中,circRNA的表达水平,如circCdr1和circular-to-linear比率与PTBP1的下调及其补偿因子PTBP2的上调高度相关(图2h)。总之,这些结果证明了circRNA的高度细胞特异性表达景观,并进一步揭示了circRNA生物发生与RBP活性之间的复杂关联,特别是在这些抑制性和兴奋性神经元中。
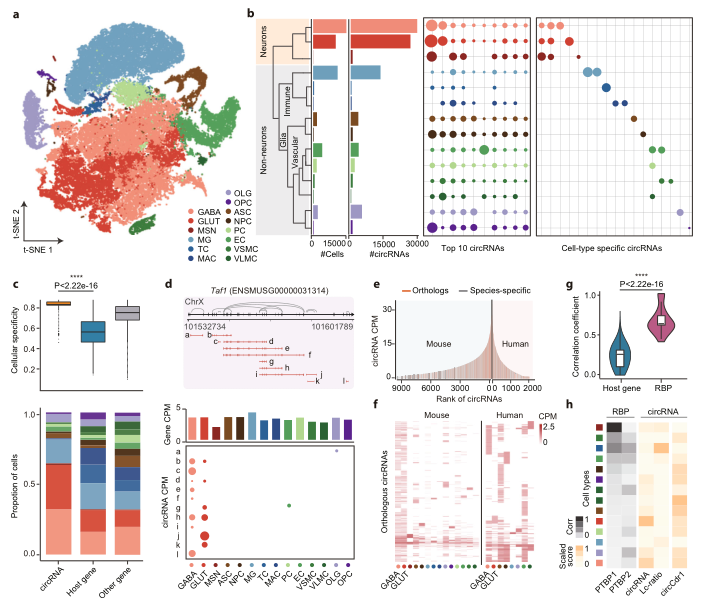
图2 抑制性和兴奋性神经元中具有丰富的circRNA
3、早期胚胎发育过程中母体和合子circRNA的动态表达
单细胞RNA测序使胚胎发育阶段的基因异质性研究成为可能,但这一过程中circRNA表达模式的变化仍需进一步探索。作者分析了11项人类和小鼠胚胎研究,其中包含来自16个从卵母细胞到早期芽的不同阶段的样本(图 3a)。在人和小鼠胚胎中分别检测到41,041和24,818个circRNA。为了揭示胚胎发育过程中circRNA之间的动态变化,计算了不同阶段circRNA表达水平之间的Pearson相关性。如图3b所示,在受精后的前3-4天观察到细胞之间的高度相关性,这与circRNA在早期胚胎发育过程中的母体效应一致。此外,从囊胚到植入胚胎的细胞表现出不同的 circRNA 表达模式,表明合子 circRNA 在囊胚期后表达。此外,在人类和小鼠样本上均观察到在发育阶段检测到的circRNA的多样性和连接率都有所增加,这也验证了这些合子circRNAs在胚胎发育过程中的积累(图3c)。考虑到在人类数据集中只收集到相对较少的细胞,下游分析只包括小鼠胚胎。为了消除随机性效应,可以在两个以上阶段检测到circRNA的表达模式绘制在图3d中。如预期的,观察到母体 circRNAs 逐渐降解,大多数其他 circRNAs 表现出阶段特异性表达谱。为进一步研究母体向合子转变过程中circRNA的动态表达变化,将样本分为四个时间点,包括全能卵裂球(TB)、第一谱系(TE/ICM)、第二谱系(EPI/PE)和植入胚胎,反映发育过程中全能性和谱系分离的变化。随后,将基因和circRNA聚类为5组。如图3e所示,簇1和簇2中的circRNA和基因在TB早期高表达,然后随着胚胎发育不断下降。相反,第3到第5簇 circRNA代表受精后特异性表达的合子circRNA。
为确定合子circRNA的激活是否是宿主基因表达的副产物,检查了circRNA与其宿主基因之间的对应关系。大部分合子circRNA(簇3中67.50%、簇4中69.2%和簇5中83.9%)是由母体表达的基因产生的,这表明这些合子circRNA在胚胎发育过程中具有独特的生物发生机制(图3h)。为进一步研究合子基因和circRNA激活过程之间的差异,计算每个簇中基因和circRNA的reads组成。仅包括在一个以上阶段中同时表达的circRNA。与发育阶段合子基因读数的温和增加相反,在图3g中观察到8个细胞阶段后合子 circRNA的急剧爆发,为母体circRNA降解和合子circRNA激活提供了令人信服的证据。例如,作者展示了两个合子和三个母体 circRNA 的不同表达模式。如图3h所示,源自Erdr1的mmu-Erdr1_0001和mmu-Erdr1_0002是一种调节细胞存活和细胞凋亡的分泌因子,在植入的胚胎中高度表达。因此,这些circRNA的高度特异性表达表明,与线性基因相比,circRNA 经历了更显著的母体到合子的转变过程。最后,对母本和合子circRNA的亲本基因进行基因本体富集分析。如图3i所示,基于微管的运动和纤毛组装在母体circRNA中富集,而剪接相关过程在合子circRNA中富集,这与发育中胚胎的极性建立和胚胎基因组激活一致。总的来说,这些结果证明了circRNA 的高度细胞特异性表达谱和合子circRNA在胚胎发育中的大量激活,这也表明了这些母体和合子circRNA 在此过程中的重要作用。
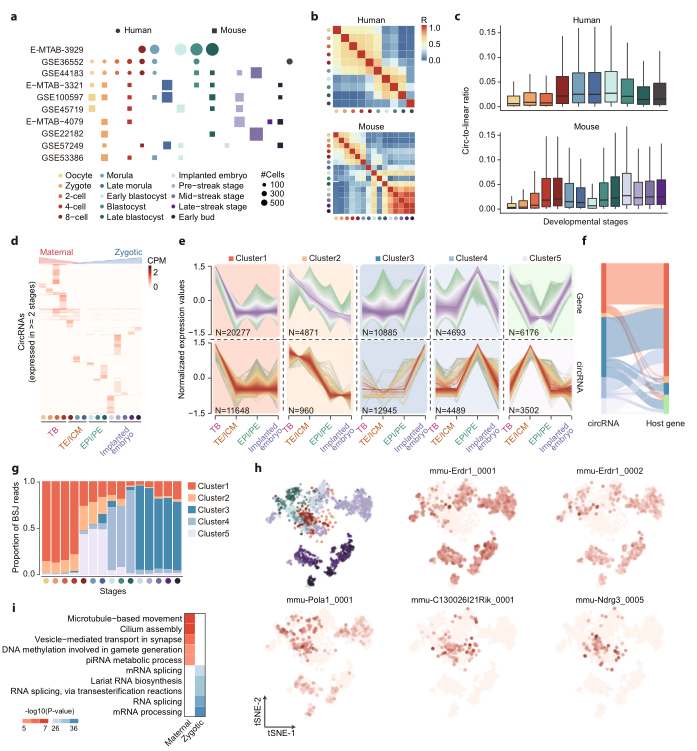
图3 母体向合子转变过程中合子circRNA 激活的解析
4、在人类乳腺癌转移中的肿瘤间和肿瘤内circRNA异质性
为分析乳腺癌肿瘤发生过程中的单细胞水平的circRNA,对26个原发性和转移性肿瘤 scRNA-seq 样本进行分析,如图4a所示。然后,进一步研究正常人群和癌人群circRNA表达水平的差异。如图4b所示,超过49.88%的正常人群和67.28%的癌人群被鉴定为上皮细胞。与之前的研究一致,非整倍体重排的肿瘤细胞在转移瘤和原发瘤中circRNAs的表达均显著降低(图4c),在大多数已鉴定的细胞类型中也观察到同样的情况(图4d)。来自预后较好的低级别(luminal A、luminal B和HER2阴性)肿瘤的正常细胞和癌细胞往往比高级别三阴性乳腺癌(TNBC)细胞表达更多的circRNA,这表明积累较少TNBC细胞中的circRNAs 具有更快的进展速度。
鉴于该队列中上皮细胞的主要数量以及EMT在肿瘤侵袭和转移中的重要作用,所以作者进一步研究了EMT期间的circRNA。首先,将所有上皮细胞聚集在一起,并进行轨迹推断分析以揭示动态细胞的分化过程(图4f)。为了更好地探索单个细胞的过渡状态,计算了EMT分数。如图4g所示,细胞轨迹结果通常相应地拟合EMT分数的增加。GO富集分析上皮细胞增殖过程在EMT评分较低的簇中富集,而细胞迁移和间充质相关过程在EMT水平较高的簇中富集。此外,计算每个簇中癌细胞的比例,并相应地观察到肿瘤细胞百分比与EMT评分之间的正相关(图4h)。最后计算每个簇中circRNA的表达水平,随着从上皮细胞(簇 1-2)到中间EMT状态(簇 3-5)的转变,circRNA 的平均表达水平相应增加(图 4i),这与EMT期间circRNA的全局激活一致。总之,作者分析了EMT期间circRNA表达的详细概况,揭示了乳腺癌患者原发性和转移性样本之间circRNA 的复杂肿瘤间和肿瘤内异质性。
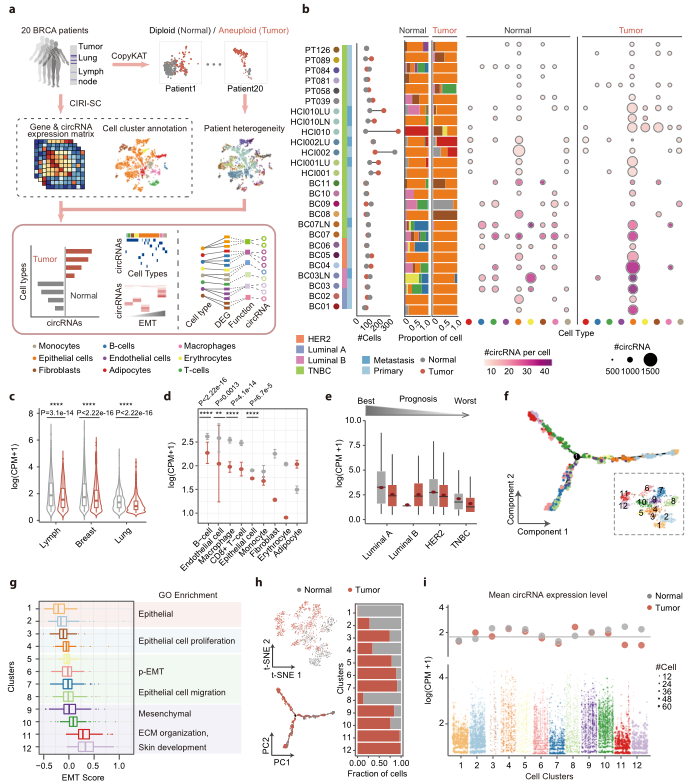
图4 乳腺癌患者正常细胞和肿瘤细胞之间circRNA的异质性
5、细胞特异性circRNA为最佳细胞类型的识别提供了的见解
基于circRNAs的高度细胞特异性,作者推测了利用circRNAs作为生物标志物来提高细胞类型的可能性。为了构建高质量的circRNA特征矩阵,研究了来自17个不同人类和小鼠组织的scRNA-seq队列以及同源的癌症样本(图5a)。在不同细胞类型和组织类型中所有的circRNA根据其表达模式被分为5类(图5b)。随后,作者总结了人和小鼠样本中circRNAs的细胞类型特异性,共享的circRNAs的关系如图5c所示。与之前研究中报道的基因表达图谱相似,circRNAs 在不同功能的细胞类型之间也表现出不同的表达簇。此外,还检测到人和小鼠细胞之间的几种直系同源细胞类型特异性circRNA,这意味着这些circRNA亚群具有保守的生物学功能。
为了验证circRNA作为细胞类型生物标志物的潜力,计算了在不同细胞类型中表达的circRNA与bulk RNA-seq数据集之间的重叠。如图5d所示,在bulk RNA-seq 数据中检测到的circRNA与细胞表达的circRNA具有高度特异性的重叠。例如,在GABAergic神经元中检测到的39.36%的circRNA也可以在正常脑样本中同时检测到。为了比较circRNA和基因作为细胞生物标志物在分析肿瘤浸润细胞中的潜能,只有在人类肿瘤样本中注释到的细胞类型被用于下游分析。之后,计算了所有表达的circRNA、来自公共数据库的标志基因和1000个随机选择基因的细胞类型特异性。值得注意的是,circRNAs的细胞类型特异性显著高于标记基因和随机对照基因,这进一步表明circRNAs作为细胞类型生物标志物的能力(图5e)。然后,使用CIBERSORT68计算癌症相关的bulk RNA-seq数据集中肿瘤浸润免疫细胞的组成,分别基于LM22基因组的标记基因和细胞类型特异性circRNA的表达(图5f)。基于circRNA和基因的反卷积结果都被整合到 scRNA-seq 队列中鉴定的10种免疫细胞类型中。随后利用对数尺度均方根误差(RMSE)评估CIBERSORT 提供的细胞特异性反卷积的结果,它代表原始标记基因表达值和推算标记基因表达值之间的偏差。如图5g所示,使用circRNA的反卷积结果具有显著更低的RMSE值,这表明circRNA估计细胞组成的效果更准确。这些结果证明了circRNA在探索肿瘤浸润性免疫细胞异质性方面作为更好的细胞类型生物标志物的适用性,也表明了这些circRNA在某些细胞类型中的重要生物学作用。
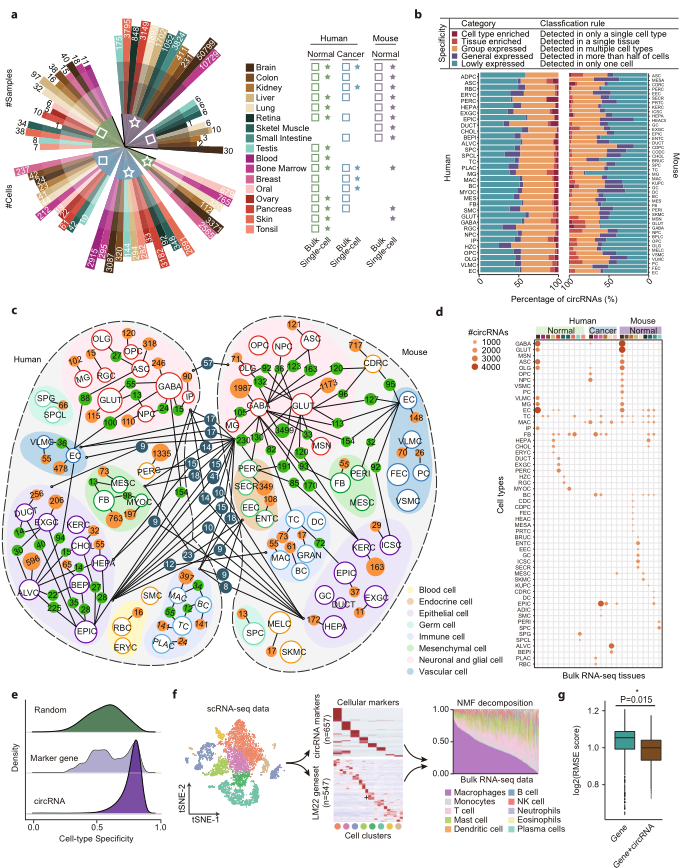
图5 探索细胞类型特异性circRNA作为细胞成分去褶积的生物标志物
作者将circRNA的细胞结构和免疫细胞中的circRNA特征矩阵集成到称为 circRNA单细胞门户(circSC)的网络服务器中。circSC提供全面的circRNA信息,包括细胞表达谱、差异表达结果以及在大量人类和小鼠细胞中鉴定的 circRNA 目录(图 6)。circSC已作为单独的模块集成到circAtlas中(http://circatlas.biols.ac.cn/),为circRNA的单细胞和bulk RNA-seq表达模式提供方便的浏览和搜索功能。作者认为该数据库可以作为探索circRNA在胚胎发育、组织分化和癌症生物发生过程中动态变化的重要资源,并为circRNA群落提供一个独特而有用的平台。
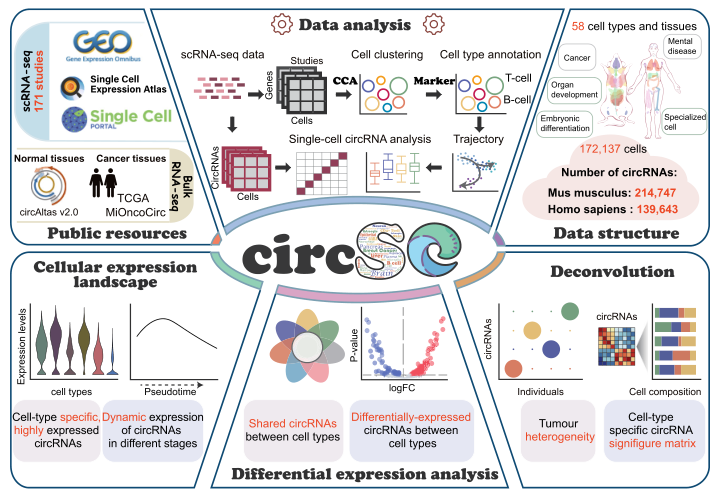
图6 circSC在线网站的建设与功能
参考文献:
Wu Wanying., Zhang Jinyang., Cao Xiaofei., Cai Zhengyi., Zhao Fangqing.(2022). Exploring the cellular landscape of circular RNAs using full-length single-cell RNA sequencing. Nat Commun, 13(1), 3242. doi:10.1038/s41467-022-30963-8