






线粒体 交联 铁死亡 在心肌病中的作用
线粒体氧化磷酸化(OXPHOS)系统缺陷引起的线粒体疾病的常见表型是心肌病和心衰。线粒体蛋白酶通过降解错误折叠的蛋白质和平衡线粒体动力学和线粒体自噬来控制线粒体的适应性。线粒体内膜(IM)蛋白酶Oma1在线粒体应激时被激活,如线粒体去极化、氧化应激、错误折叠的IM蛋白质积累、或热应激。本研究发现OMA1-DELE1介导的综合应急反应(ISR)在线粒体心肌病中具有保护作用,并将铁死亡与OXPHOS缺乏和线粒体疾病联系起来。本研究于2022年9月发表在《Cell Metabolism》IF:31.373期刊上。
技术路线如下:

主要结果如下:
为检测Oma1在线粒体OXPHOS疾病中的作用,在肌酸激酶启动子(Ckmm-Cre)的控制下,用表达Cre重组酶的小鼠繁殖Cox10fl/fl和Oma1fl/fl,从骨骼肌和心肌中敲除Cox10和Oma1基因。如图1A,心脏和骨骼肌中Cox10缺失导致小鼠早发扩张型心肌病和死亡(中位年龄31天)。Cox10−/−小鼠表现为生长迟缓,骨骼肌量进行性减少,心脏肿大,并伴有轻度结缔组织积累(图1B-E)。如预期的,观察到COX亚基和组装的COX复合体的水平急剧下降,心脏COX活性降低(图1F-G) 。通过透射电子显微镜观察到心肌组织的线粒体碎裂,线粒体超微结构改变,线粒体肿大,嵴破裂(图1H)。心脏中Cox10的缺失导致了Oma1的激活,表现为Opa1处理的增加和Opa1短形式c和e的积累(图1I)。
此外,Oma1的缺失使Cox10-/-小鼠的心脏表型恶化,而单独的Oma1-/-缺失没有明显影响。Cox10-/-Oma1-/-小鼠的寿命较短,中位生存期为 22 天(图 1A),并且表现出更严重的心肌病和纤维化病变,表明细胞死亡(图 1B-1E)。Cox10-/-小鼠中Oma1的缺失稳定了长Opa1形式并减少了线粒体碎片(图1I)。然而,在缺乏Oma1的Cox10-/-心脏中,COX 亚基和组装的COX复合物的积累没有受到影响,嵴形态也没有恢复(图 1F-1H)。这些实验揭示Oma1在体内OXPHOS 缺乏中的保护作用。

OXPHOS缺乏可通过多种机制损害各种小鼠组织和细胞模型的溶酶体功能。与此报道一致,在Cox10−/−心脏中观察到含有线粒体和膜聚集物的大型溶酶体结构(图2A-C),并观察到溶酶体标记物Lamp1和自噬底物p62/SQSTM1(简称p62)的积累(图2D-E)。与Cox10−/−心脏相比,缺陷溶酶体结构在Cox10-/-Oma1-/-心脏中进一步积累,并且在Oma1缺失后Lamp1和p62的稳态水平显著增加(图2F-G)。这些结果表明,在Cox10−/−缺陷的心肌细胞中,线粒体功能障碍导致溶酶体缺陷,且在缺少Oma1的情况下进一步恶化。
由于观察到pS6和p4E-BP1、mTORC1底物在Cox10−/−和Cox10-/-Oma1-/-心脏中积累(图-2H-I),所以进一步检查了用雷帕霉素抑制mTORC1后是否会延长小鼠的寿命。结果显示雷帕霉素抑制Cox10−/−和Cox10-/-Oma1-/-心脏S6磷酸化(图2H-I)。然而,用雷帕霉素喂养不影响小鼠的存活(图2J)、线粒体超微结构(图 2K)、p62和Lamp1的积累(图2H-I)。 因此,得出结论,增加mTORC1信号传导不会导致这些小鼠的溶酶体缺陷,并且Oma1通过另一种机制延长Cox10−/−小鼠的寿命。
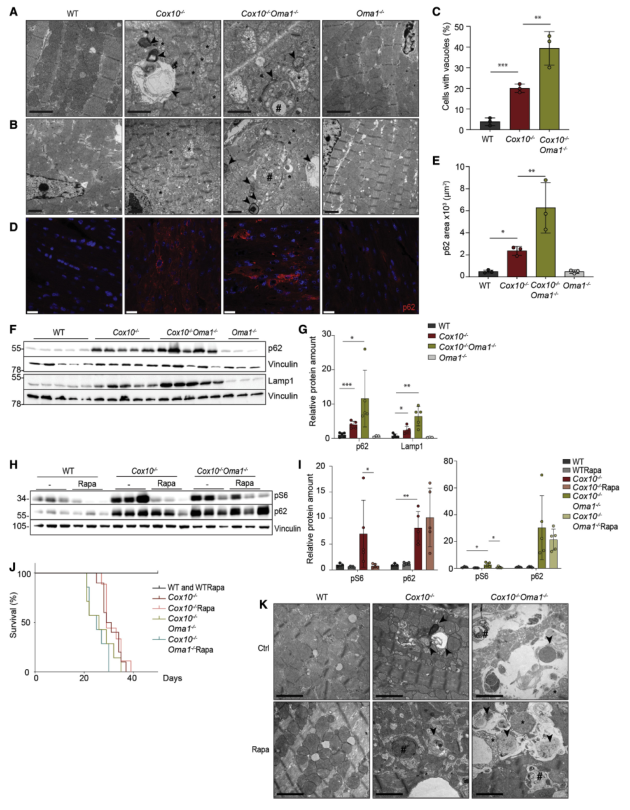
图2 线粒体心肌病中Oma1缺失导致溶酶体缺陷和p62积累
3、Oma1激活Cox10−/−小鼠心脏的ISR
对4周大野生型、Cox10−/−、Oma1−/−、Cox10−/− Oma1−/−小鼠心脏组织进行测序并对差异表达基因进行KEGG分析。结果鉴定到Nrf2信号通路是Cox10−/−心脏中最显著诱导的通路(图3A)。根据靶基因表达,p53、Atf4和Nrf2是在Cox10−/−心脏中被预测为最显著改变的转录因子(图3B)。比较Cox10−/−和Cox10−/− Oma1−/−小鼠心脏的转录组,发现Atf4是变化最显著的转录因子(图3C)。根据文献报道,作为ISR的一部分,Atf4和其它转录因子一起介导eIF2α信号通路可响应不同的线粒体损伤。与Cox10−/− Oma1−/−心脏的ISR受损一致,在Cox10−/− Oma1−/−小鼠心脏中,Atf4靶基因Fgf21、Pycr1和Mthfd2的表达明显低于Cox10−/−小鼠(图3D)。对上述4种小鼠的TMT无偏蛋白组分析证实了以上发现(图3E)。免疫印迹证实,与Cox10−/−心脏相比,Cox10−/− Oma1−/−心脏中Atf4和eIF2α磷酸化水平降低(图3F-G)。综上,在心脏OXPHOS缺乏的反应中,Oma1是诱导Atf4介导ISR所必需的。
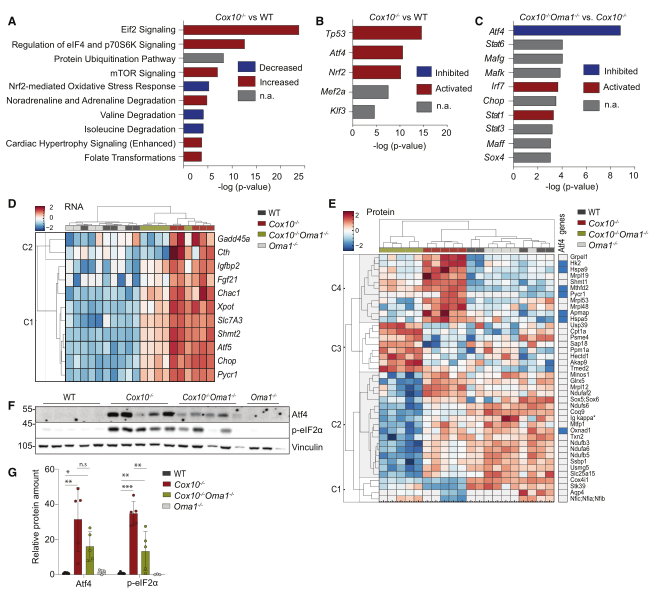
图3 Cox10−/−小鼠心肌表现出Nrf2和p53激活以及Oma1和Dele1介导的Atf4信号通路
4、Cox10−/−小鼠中ISR诱导是Dele1依赖的
作者在此前的实验中为探究Oma1作用ISR激活的底物,比较了4种小鼠的线粒体蛋白组,分析发现Oma1促进了Cox10−/−心脏中可能未组装的COX亚基的蛋白水解,,其蛋白水解可能介导ISR激活,但没有发现候选Oma1底物。虽然在心脏中表达,但作者通过质谱没有检测到Dele1,Dele1已被确定为Oma1底物和Atf4依赖性线粒体应激信号的关键成分。因此,构建Dele1−/−小鼠,并将其与心脏和肌肉特异性Cox10−/−小鼠杂交,以研究Dele1在体内对ISR信号的作用。敲除Dele1对小鼠的生存没有广泛影响,直到1岁时,在Dele1缺失的情况下,小鼠都没有表现出任何扩张表型,然而,当同时缺失Cox10时,Dele1消融加重了Cox10−/−小鼠的表型,这些小鼠在约4周龄时死亡(图4A)。与Cox10−/−小鼠相比,Cox10−/− Dele1−/−小鼠的体重进一步降低,心脏/体重比进一步增加,且心脏进一步扩张(图4B-D),但不影响COX活性(图4E)。在缺少Cox10的情况下,Dele1的缺失完全消除了Atf4和peIF2α的积累(图4F-G),降低了ISR靶基因的表达(图4H)。这些结果表明,Dele1的缺失与Oma1缺失类似同样影响心脏特异性Cox10−/−小鼠的表型。ISR在Cox10−/−小鼠心脏中沿Oma1-Dele1轴被激活(图4I),这与小鼠寿命延长有关。

图4 Cox10−/−小鼠心脏Dele1缺失恶化线粒体心肌病并损伤ISR
5、Oma1介导的ISR反应支持心脏的谷胱甘肽代谢
为确定OXPHOS缺乏时ISR受损的代谢后果,在4种小鼠心脏中进行靶向代谢组学研究(图5A-B)。观察到Cox10−/−心脏中单碳代谢中间体如丝氨酸、甘氨酸和蛋氨酸的积累,以及天冬酰胺、丙氨酸、支链氨基酸和脯氨酸的积累,Oma1的缺失没有影响这些代谢物的稳态水平,也没有强烈影响它们在Cox10−/−心脏中的积累(图5A)。完全相反,Cox10−/−心脏中GSH的还原形式的积累在Cox10−/−Oma1−/−心脏中显著增加(图5B),这表明Oma1在OXPHOS缺乏时调节GSH水平。GSH代谢与单碳代谢密切相关,由Atf4和Nrf2共同调控(图5C)。与Oma1缺失时ISR受损一致,Cox10−/−心脏中参与GSH代谢的酶的表达增加,而与Cox10−/−心脏相比,Cox10−/−Oma1−/−心脏中几种酶的表达显著降低(图5D)。蛋白质组显示,许多这些酶在 Cox10 缺陷的心脏中的积累和在没有Oma1的情况下有较低的稳态水平(图5E)。这些蛋白质包括GSH依赖的脂质过氧化物酶Gpx4,GSH降解酶Chac1和Chop1,它们的积累通过对心脏组织的WB进行监测(图5F-G)。总之,得出结论,Oma1介导的ISR支持Cox10−/−心脏中的GSH代谢。

图5 Oma1介导的ISR支持线粒体心肌病的谷胱甘肽代谢
6、Cox10−/−Oma1−/−心脏具有铁死亡特征
对心脏RNA-seq数据中与细胞死亡相关的基因进行无监督聚类,发现这些基因在Cox10−/−和Cox10−/−Oma1−/−心脏中有不同的调控,包括促凋亡蛋白Chop和Chac1(图6A)。心脏切片的TUNEL染色检测到Cox10−/−中TUNEL阳性心肌细胞比Cox10−/−Oma1−/−中更多(图6B-C)。然而,检测到的TUNEL阳性细胞数量较少不能解释Cox10−/−小鼠心肌病的严重程度,也不可能解释Cox10−/−Oma1−/−小鼠的加重表型。此外,在Cox10−/−Oma1−/−心脏中存在炎症反应(图6D)。结合该结果以及GSH和铁死亡有关等,因而检测了MDA的积累。结果发现在Cox10−/−中能检测到MDA的积累但是在Cox10−/−Oma1−/−心脏中积累显著增加(图6E,H)。透射电子显微镜观察到脂质过氧化旋涡的形成(图6F)。此外,染色也观察到Cox10−/−Dele1−/−小鼠心脏中脂质过氧化的形成。证实这种死亡与铁死亡有关。此外,与Cox10−/−Oma1−/−小鼠相似(图5E-5G),Cox10−/−心脏中Dele1的丢失使Gpx4蛋白水平降低到野生型水平(图6J)。因此,Oma1或Dele1的缺失,消除 Cox10-/-心脏中的ISR,与Gpx4蛋白水平降低和脂质过氧化增加有关,表明 Oma1-Dele1-Atf4 信号轴可防止OXPHOS缺陷心脏中的铁死亡(图6K)。

图6 Cox10−/−小鼠心脏表现出Oma1依赖的铁死亡特征和炎症
7、依赖于Oma1的ISR在体外抵御铁死亡
为明确ISR信号对铁死亡的抵抗作用,分离4种小鼠的MEFs细胞。观察到与WT、Cox10−/−、Oma1−/−细胞比较,Cox10−/−Oma1−/−细胞的存活率显著下降(图7A-B),但该情况在铁死亡抑制剂Fer-1存在的情况下显著抑制(图7C),在铁死亡诱导剂erastin存在的情况下加剧(图7D-E)。提示Oma1保护Cox10−/−MEFs细胞抵御铁死亡。
与体内结果类似,观察到Atf4,GPX4,Chop在Cox10−/−而非Cox10−/−Oma1−/− MEFs细胞中积累(图7F),同时,Atf4靶基因的Oma1依赖性表达增加,包括参与GSH代谢的各种酶(图7G)。这些基因在Cox10−/−积累也依赖于Dele1(图7H)。对这4种MEFs细胞的代谢组分析揭示了半胱氨酸、胱硫氨酸和谷胱甘肽在Cox10−/−而非Cox10−/−Oma1−/− MEFs细胞中积累(图7I)。对Cox10−/−细胞施以eIF2α磷酸化和ISR信号通路抑制剂ISRIB处理。短时间用ISRIB处理增加野生型细胞对erastin的敏感性,而Cox10−/−的存活在这些条件下不受影响,在ISRIB长时间抑制ISR后,erastin诱导的Cox10−/−细胞铁死亡增加(图7J)。同样,Atf4或Dele1缺失增加了Cox10−/−细胞对erastin诱导的铁死亡的易感性(图7K)。这些证明Oma1-Dele1介导的ISR对铁死亡有抵御作用。
此外,图7L-M进一步证明Oma1支持细胞对硒的利用并促进Gpx4的积累。因此,Oma1介导的ISR增加了转硫作用途径酶的转录,从而支持硒的利用,从而促进Gpx4的积累和对脂质过氧化和铁死亡的抵抗力(图7N)。

图7 ISR沿Oma1-Dele1-Atf4轴激活保护Cox10−/− MEFs抵抗铁死亡
总之,如图8所示,Cox10−/−心脏中的 OXPHOS 缺乏会导致心肌病并引发沿 Oma1-Dele1-Atf4 轴的综合应激反应。Oma1依赖的应激信号可保护谷胱甘肽代谢和Gpx4积累,以限制脂质过氧化、抑制铁死亡和延缓心肌病。
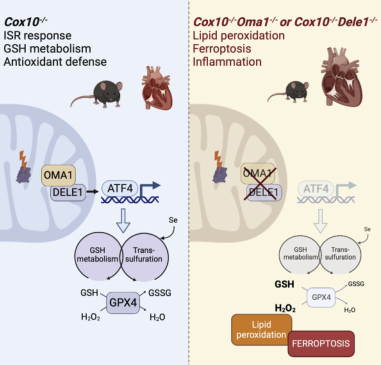
图8 图形摘要
参考文献:
Ahola Sofia., Rivera Mejías Pablo., Hermans Steffen., Chandragiri Srikanth., Giavalisco Patrick., Nolte Hendrik., Langer Thomas.(2022). OMA1-mediated integrated stress response protects against ferroptosis in mitochondrial cardiomyopathy. Cell Metab, undefined (undefined), undefined. doi:10.1016/j.cmet.2022.08.017