






线粒体联动铁死亡保卫心脏的新策略
细胞铁死亡是由铁依赖性脂质过氧化作用驱动的调节性细胞死亡的一种形式。线粒体在细胞铁死亡中发挥着重要的作用,也与一系列疾病显著相关。线粒体GSH(mitoGSH)库的过度耗竭或氧化关联到多种病理生理条件。本研究发现FUNDC2通过调控mitoGSH来调节铁死亡应激。研究强调通过阻断铁死亡和改善mitoGSH库和功能来保护DOX诱导的心肌病。这可能成为一种保护心脏的新策略。本研究于2022年9月发表在《PNAS》IF:12.779期刊上。
技术路线:
主要研究结果:
作者首先通过WB分析筛选主要器官中FUNDC2的表达情况,发现FUNDC2在心脏中高表达,暗示它的潜在功能(图1A)。也发现,当个体完全丢失FUNDC2蛋白后(图1B),其后代的表型符合孟德尔定理而且正常生长。另外,FUNDC2敲除(FUNDC2-KO)的成年小鼠拥有正常的体重和健康的心脏功能(图1C-G)。接着作者给野生型小鼠和FUNDC2-KO小鼠分别注射一剂10 mg/kg DOX(生理盐水作为对照),结果发现与野生型小鼠相比,FUNDC2-KO小鼠的体重和心脏重量显著显著减少(图1C)。经胸壁超声心动图通过测量左心室射血分数(LVEF)和左心室缩短率(LVFS)用来评估心脏功能(图1D)。注射生理盐水出的WT型小鼠和FUNDC2-KO小鼠在LVEP和LVFS上均没有明显的变化,但是与空白对照WT型小鼠相比,用DOX处理过FUNDC2-KO小鼠的心脏功能被保护地更好(图1E)。用DOX处理WT小鼠的心房利钠肽(Anp)、脑钠肽(Bnp)、肌球蛋白7(Myh7)等生物标志物的mRNA水平都显著上调,而盐处理组没有差异(图1F)。这些心力衰竭生物标志物的mRNA水平也支持这一观点。Masson ' s Trichrome染色表明FUNDC2的敲除可以防止DOX诱导的心脏纤维化(图1G)。这些结果表明丧失FUNDC2保护小鼠免受DOX诱导的心肌病。
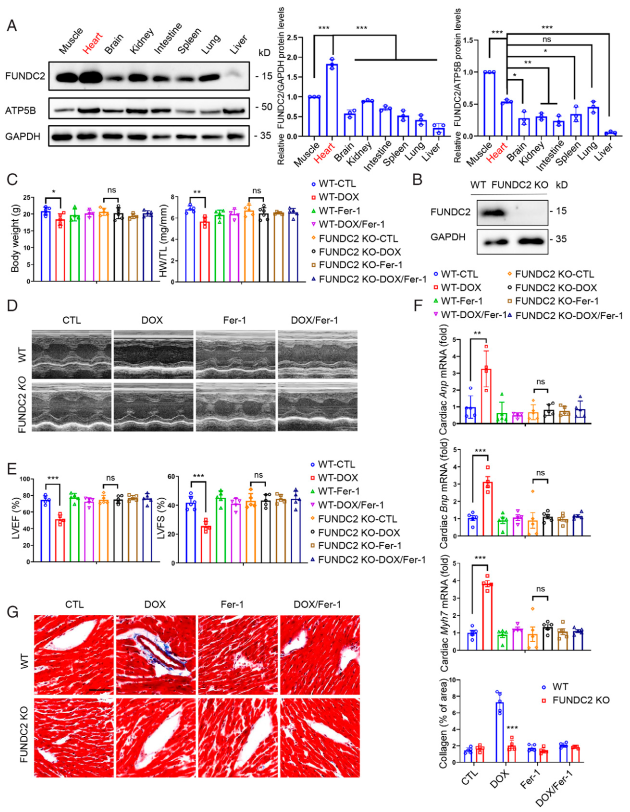
图1敲除FUNDC2可改善DOX诱导的心肌病
作者推测FUNDC2通过调节细胞铁死亡影响DOX诱导的心脏中毒。作者首先验证Fer - 1预处理对WT和FUNDC2-KO两种小鼠心脏损伤都有很强的保护作用(图1C-G)。接着进一步探索FUNDC2在DOX诱导的铁死亡中的作用。结果发现,DOX诱导WT小鼠心脏的Ptgs2 mRNA表达显著上调近3倍,而Fer - 1预处理后上调作用被抑制(图2A)。然而,在KO小鼠中没有观察到Ptgs2 mRNA表达的变化(图2A)。
作者进一步检测脂质过氧化的生物标志物,4 - 羟基壬烯醛(4-HNE)的水平,这也是体内铁死亡的关键指标。结果发现4 - HNE水平在DOX处理的WT小鼠心脏组织中显著增加(图2B-C),但在FUNDC2 - KO小鼠中没有。作者还分析脂质过氧化产物丙二醛(MDA)的水平,发现其上调只能在WT小鼠中检测到,而在FUNDC2 - KO小鼠中检测不到(图2D,左)。作者随后分离心脏组织的亚细胞组分,发现DOX处理增强线粒体中的MDA水平,但不在细胞质中(图2D,中间,右)。值得注意的是,MDA只在野生型小鼠中增加,不在FUNDC2 - KO小鼠中增加(图2D,中间,右)。作者的结果与DOX处理特异性增强线粒体脂质过氧化的观察一致。相反,FUNDC2缺失可以阻止DOX诱导的脂质过氧化增加。作者进一步使用电子显微镜检查线粒体形态,发现DOX处理心脏中的线粒体破裂,嵴扭曲,嵴密度降低(图2E)。相比之下,FUNDC2的缺失在很大程度上阻止这种线粒体损伤。最后,Fer - 1强烈抑制DOX诱导的线粒体变形,暗示DOX -线粒体-铁死亡信号轴。
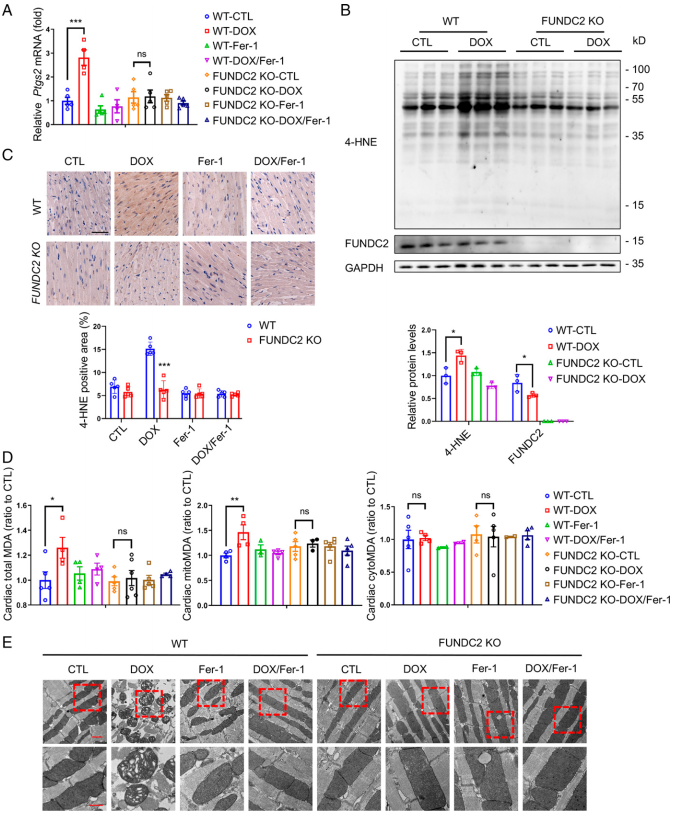
图2敲除FUNDC2缓和DOX诱导的铁死亡。
3、FUNDC2 - KO抑制Erastin诱导的细胞死亡
GSH代谢处于氧化还原代谢和铁死亡的中心。作者检测心脏组织中总GSH / 谷胱甘肽二硫化物(GSSG)比值和GSH水平,发现FUNDC2 - KO心肌中GSH / GSSG比值和GSH水平远高于WT组(图3A)。由于FUNDC2是一种线粒体蛋白,作者试图确定FUNDC2是否调节线粒体GSH。事实上,FUNDC2 - KO心脏中线粒体GSH / GSSG比值和GSH水平高于WT组(图3A)。值得注意的是,在WT心脏中,总的和线粒体的GSH / GSSG比值和GSH水平都被DOX处理降低到50 %,但在KO心脏中保存(图3A)。这些数据表明,在DOX诱导的心脏中毒中,高GSH水平,特别是mitoGSH,可能具有心脏保护作用。
为进一步理解FUNDC2管理mitoGSH的分子机制,作者用WT和FUNDC2-KO MEF细胞进行体外研究。与体内结果一致,FUNDC2 - KO MEF细胞中线粒体和胞浆GSH / GSSG比值和GSH (cytoGSH)水平显著高于WT MEF细胞(图3B)。Erastin处理降低WT和FUNDC2 - KO MEF细胞的GSH / GSSG比值和GSH水平,但FUNDC2 - KO MEF细胞的GSH / GSSG比值和GSH水平在所有条件下均高于WT MEF细胞(图3B)。GSH水平与erastin诱导的细胞死亡相关(图3C),表明GSH在铁死亡中的功能重要性。使用脂质过氧化的荧光探针C11 - BODIPY通过流式细胞术检测脂质ROS的积累,证明FUNDC2的缺失阻止erastin诱导的脂质ROS积累(图3D)。DOX诱导的铁死亡也在MEF细胞中得到证实。
为进一步证实FUNDC2通过mitoGSH促进铁死亡,作者用mitoGSH处理MEF细胞,并用一种特定的mitoGSH消耗化学物质mitoCDNB选择性损耗mitoGSH。mitoCDNB预处理降低线粒体GSH / GSSG比值和GSH水平,erastin处理后进一步降低(图3E)。mitoCDNB处理有效增强erastin诱导的WT细胞和FUNDC2- KO MEF细胞死亡和脂质ROS积累(图3F-H)。这些结果表明mitoGSH耗竭确实增加erastin诱导的细胞死亡。
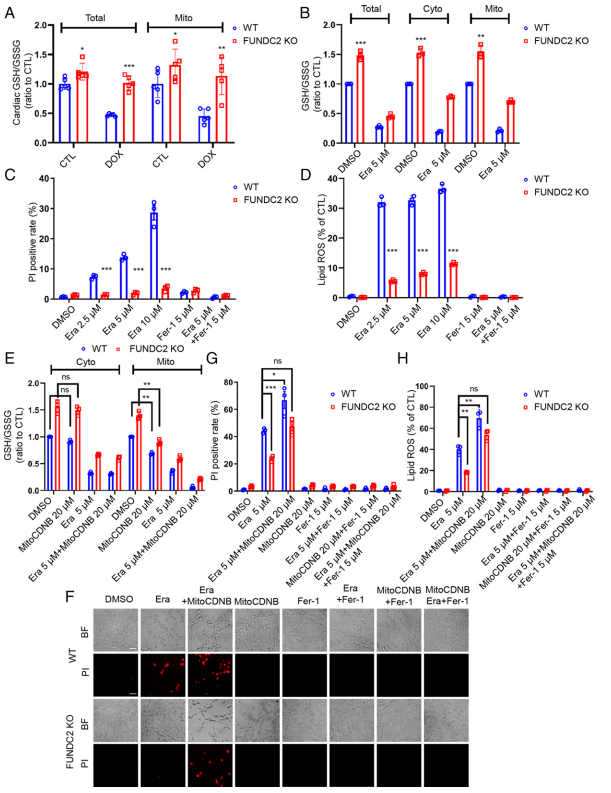
图3FUNDC2-KO抑制但mitoGSH耗竭增加erastin诱导的细胞死亡。
作者研究FUNDC2与SLC25A11之间的相互作用是否在铁死亡中发挥功能。,作者首先以scramble shRNA作为对照,在WT和FUNDC2 - KO MEF细胞中稳定敲低SLC25A11(图4A),发现SLC25A11缺失降低WT和FUNDC2 - KO MEF细胞中线粒体GSH / GSSG比率和GSH水平,而对胞浆GSH / GSSG比率和GSH水平无明显影响(图4B)。SLC25A11 KD也增强erastin诱导的铁死亡,无论FUNDC2水平如何,细胞活力,细胞死亡(图4C-E)和脂质ROS积累都证明了这一点(图4F)。这些数据表明SLC25A11对于细胞抵抗铁死亡是必需的。
作者进一步探索在SLC25A11中获得功能是否可以防止erastin诱导的铁死亡。将编码载体或Flag - SLC25A11的质粒稳定转染到WT和FUNDC2 - KO MEF细胞中(图4G),发现在WT和FUNDC2 - KO MEF细胞中,无论是否用erastin处理,SLC25A11过表达均增加线粒体GSH / GSSG比值和GSH水平(图4H)。因此,过表达SLC25A11足以抑制erastin诱导的细胞死亡(图4I-K)和脂质ROS积累(图4L)。这些结果表明SLC25A11可以对抗FUNDC2依赖性的铁死亡。
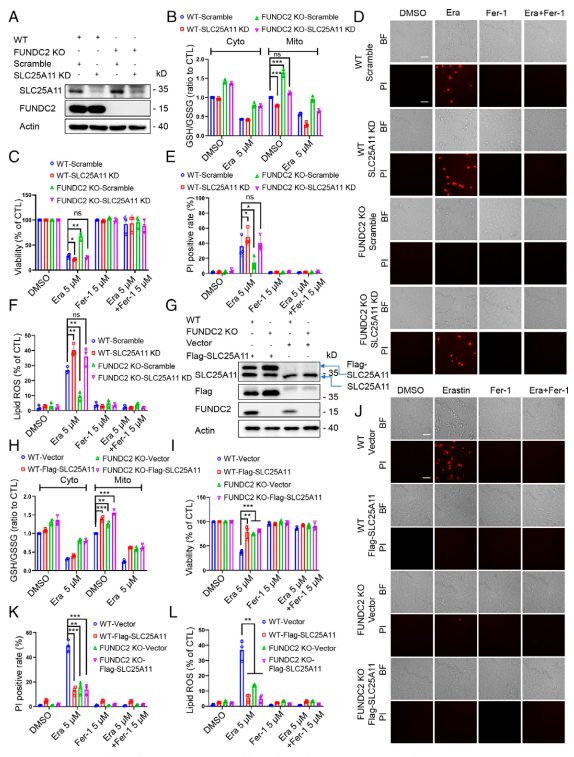
图4 FUNDC2–SLC25A11轴调节铁死亡
在这里作者探究FUNDC2如何调控SLC25A11。通过WB检测,作者发现FUNDC2-KO可以增加SLC25A11水平,并阻断erastin诱导的GPX4和线粒体蛋白如TIM23和TOM20在MEF细胞中的减少(图5A)。此外,FUNDC2 - KO小鼠心脏组织中SLC25A11水平高于WT小鼠(图5B)。没有观察到SLC25A10的影响,另一个SLC25家族成员(图5A-B)。尽管在WT和FUNDC2 - KO小鼠中,线粒体蛋白和GPX4在响应DOX时均下调,但FUNDC2 - KO缓解下调的程度(图5B)。然后作者测试在铁死亡过程中FUNDC2和SLC25A11之间的相互作用是否增强。通过免疫共沉淀实验,结果发现erastin处理增强FUNDC2和SLC25A11之间的相互作用(图5C)。类似的结果在体内心脏组织中得到重现(图5D)。
因为许多线粒体蛋白需要组装成寡聚体结构才发挥其功能,作者通过凝胶电泳检测SLC25A11的寡聚体状态。如图5E所示,在MEF细胞中,用温和的非变性洗涤剂Nonident P - 40裂解线粒体时,SLC25A11蛋白以二聚体形式存在;而在强变性洗涤剂十二烷基硫酸钠(SDS)裂解时,只能观察到SLC25A11的单体形式。无论erastin处理与否,FUNDC2-KO MEF细胞中SLC25A11的二聚体和单体蛋白水平均高于WT MEF细胞(图5E)。同样,在心脏组织和它们在体内的线粒体部分中也可以观察到差异,尽管程度相对较小(图5F)。这些结果表明FUNDC2通过调节SLC25A11的稳定性和二聚化来调节其功能。
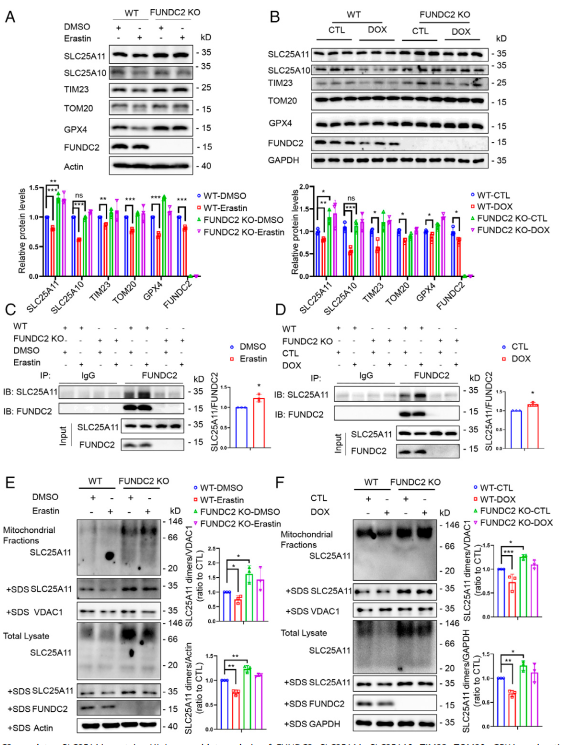
图5 FUNDC2调控SLC25A11蛋白
结论
本研究结果表明线粒体FUNDC2对于DOX诱导的心肌病是重要的。敲除FUNDC2在很大程度上预防DOX诱导的心肌病。进一步研究发现,FUNDC2调控mitoGSH以及DOX诱导的铁死亡和心脏损伤需要稳定的GPX4和SLC25A11。也说明线粒体定位的GPX4抑制脂质过氧化的产生,并防止DOX诱导的铁死亡。本研究强调通过阻断铁死亡和改善mitoGSH库和功能来保护DOX诱导的心肌病的策略。
参考文献
Ta N, Qu C, Wu H, Zhang D, Sun T, Li Y, Wang J, Wang X, Tang T, Chen Q, Liu L. (2022). Mitochondrial outer membrane protein FUNDC2 promotes ferroptosis and contributes to doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci USA. 119(36):e2117396119. doi: 10.1073/pnas.2117396119.