






新发现!转录组修饰与细胞自噬间的沟通
自噬对于维持细胞能量稳态和细胞适应营养缺乏至关重要,而调节自噬的营养传感器在此前已有报道。然而,像m6A这样的转录组修饰在调节饥饿诱导的自噬中的作用尚不清楚。本研究中,展示了m6A读取器YTHDF3对诱导细胞自噬的作用。m6A修饰上调促进自噬形成和营养缺乏时溶酶体溶解。METTL3耗尽导致m6A修饰功能缺失,抑制YTHDF3介导的自噬通量。YTHDF3通过识别FOXO3 mRNA停止密码子周围的m6A修饰位点来促进自噬。YTHDF3还会招募eIF3a和eIF4B来促进FOXO3的翻译,进而启动自噬。本研究于2022年10月4日发表在期刊《Nature communications》上,IF:17.694。
技术路线:

主要研究结果:
1、YTHDF3上调是自噬诱导所必需的
为识别参与自噬的潜在的转录因子,作者进行了蛋白质组学分析,筛选小鼠胚胎成纤维细胞(Mouse embryonic fibroblasts,MEFs)中因营养缺乏而上调的蛋白质。作者发现m6A读取器YTHDF3的水平显著增加(图1a),并用WB验证了它的上调。在营养匮乏期间,YTHDF3水平显著上升的同时,其他的YTH家族蛋白没有明显变化(图1b)。这一发现与最近报道的缺氧YTHDF1上调表达有所不同。
分析是否YTHDF3诱导与自噬体的出现密切相关,作者用两种不同的shRNA表达慢病毒敲低MEFs中YTHDF3。分析LC3-II存在或不存在溶酶体抑制剂bafilomycin A1(Baf.A1),结果发现沉默YTHDF3自噬体通量受损。作为自噬的另外一个指标,p62是一种自噬载体蛋白,能在自噬溶酶体中降解。而在对照细胞中,营养饥饿诱导p62降解,YTHDF3消融导致p62积累(图1c),也表明自噬通量减少。WB分析显示,与KD对照相比,YTHDF3恢复的营养缺乏MEF内源性LC3-II和p62降解水平均显著增加(图1d),证明YTHDF3挽救了shRNA诱导的自噬失活。YTHDF3的缺失持续且显著降低了胞质中GFP-LC3点状聚集(图1e-f),而这种缺陷可以通过重新表达YTHDF3来挽救(图1g-h)。此外,还发现YTHDF3过表达显著增强自噬标志物的表达,降低p62水平(图1i),表明YTHDF3不仅是维持生理性自噬所必需的,还介导了自噬增强。这一发现也得到了GFP-LC3实验的证实,该实验表明通过异位表达YTHDF3,细胞质中的GFP-LC3斑点数量增加(图1j-k)。透射电镜(TEM)证实YTHDF3确实导致自噬小体和自溶小体数量大幅减少(图1l)。这些数据表明YTHDF3是一个正调控因子,是在营养缺乏时诱导自噬所必需的。
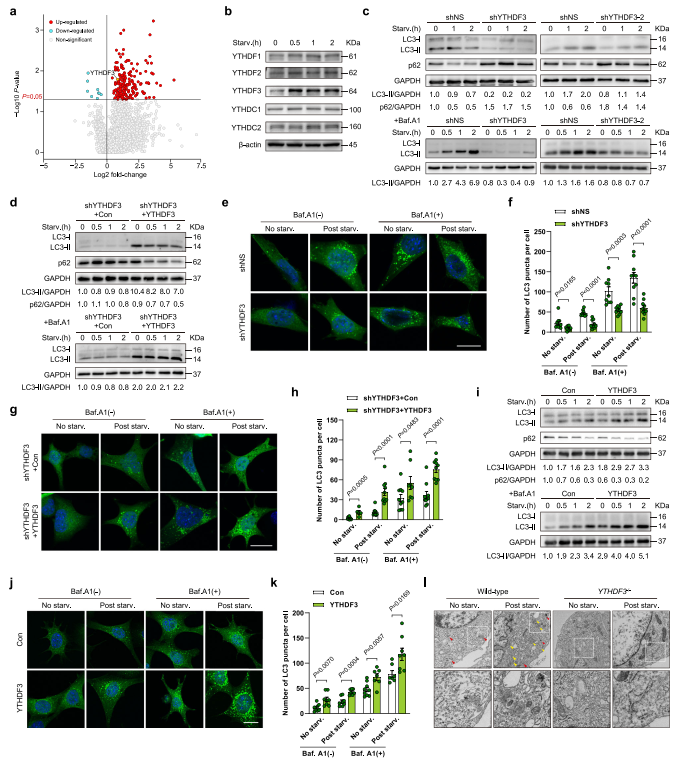
图1 YTHDF3上调是自噬诱导所必需的
2、YTHDF3衰竭损害了自噬小体的形成和溶酶体的功能,mTORC1和AMPK信号不受YTHDF3的影响
作者进一步研究YTHDF3 KD细胞中自噬的哪个步骤被中断。通过使用荧光标记的串联mCherry-GFP-LC3报告蛋白,测量自噬体和自溶酶体的丰度。在营养匮乏的条件下,YTHDF3的损失使非酸性植物(mCherry+GFP+)和酸性(mCherry+GFP−)点状数量急剧减少,说明明自噬小体的形成和自噬通量均严重受损(图2a-b)。
在哺乳动物中,自噬通过ULK1复合物的刺激而启动。当ULK1复合物被激活时,将III PtdIns3K复合物的组分磷酸化,在内质网(ER)膜上形成特化的Ptdins3P enated丰富子结构域。为探测YTHDF3是否有助于早期细胞自噬的形成。作者发现敲低YTHDF3抑制饥饿诱导的ULK1和ATG13的点形成(图2c-d),表示UKL1复合体向吞噬体起始位点的易位受到了影响。与ATG13一致,在饥饿条件下敲低YTHDF3使ATG14点的形成和PtdIns3P结合蛋白DFCP1显著减少(图2c-d)。这些结果说明YTHDF3耗尽损害早期的自噬体形成,包括起始和成核。作者选择Acridine orange(AO)作为染色剂(绿),用Baf.A1处理的细胞作为对照组,分析YTHDF3是否通过同时影响溶酶体活性来调节自噬通量。结果显示YTHDF3-KD细胞红绿荧光强度比降低(R/GFIR)(图2e),说明沉默YTHDF3可以增加溶酶体pH值。LysoTracker Red staining也证实了这一结果(图2f-g)。接下来,作者调查沉默YTHDF3是否影响溶酶体的水解功能。作者将DQ-Red BSA载入对照组和YTHDF3 KD细胞中,DQ-Red BSA在被溶酶体蛋白酶降解时释放荧光单体。YTHDF3 KD细胞中红色荧光减弱,表明细胞内蛋白水解减少(图2h-i)。因为不同的内吞率可能会影响DQ-Red BSA贩运到溶酶体中,作者使用cell-permeant Magic Red测量组蛋白酶B活性。结果显示YTHDF3 KD细胞只具有较低的Magic Red(图2j-k),提示溶酶体组织蛋白酶活性受损。这些结果表明YTHDF3耗尽损害自噬体的形成,导致溶酶体功能障碍。
基于上述结论,作者考虑YTHDF3是否影响上游mTORC1和AMPK信号的活性。作者通过测定mTOR在S2448和mTORC1下游靶点的磷酸化状态来分析mTORC1活性,包括p70S6K在T389和4E-BP1在T37/46。在对照组和YTHDF3 KD细胞中,mTOR S2448、p70S6K T389和4E-BP1 T37/46的磷酸化水平在营养缺乏时显著降低(图2l)。作者通过测量AMPK α在T172和AMPK底物RAPTOR在S792的磷酸化水平来评估YTHDF3是否调节AMPK活性。AMPK α T172和RAPTOR S792的磷酸化水平在营养饥饿时显著增加,而YTHDF3 KD细胞中的变化模式与对照组细胞相同(图2m)。这些数据表明,mTORC1和AMPK信号不受YTHDF3的影响。

图2 YTHDF3衰竭损害了自噬小体的形成和溶酶体的功能
3、YTHDF3需要METTL3介导的m6A修饰来促进自噬
为评估自噬过程中m6A的变化,作者使用一种识别m6A修饰核酸的抗体进行了免疫荧光分析。研究发现在营养缺乏时m6A信号于细胞质中积累(图3a-b)。同时,YTHDF3也在这一过程中积累(图3a-b)。用oligo(dT)纯化的Poly(A)+ RNA在营养缺乏时m6A修饰显著增加(图3c),提示mRNA的m6A水平在自噬诱导过程中升高。
作者希望鉴别了负责mRNA的m6A超甲基化的m6A编码器,发现只有METTL3在营养匮乏条件下显著诱导(图3d)。敲低METTL3降低自噬诱导过程中mRNA的高甲基化(图3e)。这说明METTL3在营养匮乏条件下对m6A的诱导是必需的。
作者观察到d3-m6A与RNA探针的摩尔比在饥饿后6小时下降(图3f),表明METTL3的催化活性下降。有趣的是,METTL3蛋白水平在长时间饥饿中没有显著变化(图3g)。作者推测这可能是细胞在营养耗竭状态下的一种代偿机制。研究使用两种不同的shMETTL3慢病毒感染过表达YTHDF3的MEFs,发现营养饥饿诱导的LC3-II积累和p62降解显著减弱(图3h)。是否METTL3对ythdf3促进自噬的作用依赖于其m6A催化活性,作者将野生型或催化突变型METTL3重新引入到METTL3沉默的细胞中,并表明野生型METTL3,而不是催化突变型,可以挽救自噬缺陷METTL3沉默细胞(图3i)。与这些结果一致的是,一项mCherry-GFP - LC3串联报告实验表明,在过表达YTHDF3的MEFs中,营养缺乏时,METTL3的损耗会影响自噬体和自溶酶体的生成(图3j-k),并且野生型METTL3可以恢复这种功能失活,而催化突变体则不能(图3l-m)。总的来说,这些数据表明YTHDF3需要METTL3介导的m6A修饰来促进自噬。

图3 YTHDF3需要METTL3介导的m6A修饰来促进自噬
4、受抑制的RPS27a-METTL3相互作用使METTL3在饥饿下稳定
为测试METTL3是否参与转录调控,作者首次分析营养缺乏状况下,METTL3 mRNA丰度。研究结果显示在营养匮乏的条件下,METTL3 mRNA丰度小幅度增加(图4a)。然后作者检测营养饥饿是否影响METTL3 mRNA的稳态,没有观察到显著差异(图4b)。但METTL3蛋白营养饥饿时上调在一个经典的蛋白酶抑制剂MG132的治疗下完全消除了(图4c)。这表明营养缺陷下METTL3是蛋白质稳定性的主要控制因子。环己酰亚胺(CHX)的chase实验证实了这一观点,揭示了饥饿状况减弱METTL3蛋白的降解(图4d)。此外,作者还发现营养饥饿显著降低METTL3泛素化(图4e)。为识别与METTL3相互作用的潜在调控因子,并解释其在饥饿反应中去泛素化的原因,作者在MEFs中过表达Flag标记的METTL3,并将抗Flag免疫沉淀物进行质谱分析。令人惊喜的是,作者发现了6个泛素化相关蛋白与METTL3共化,包括RPS27a,RPL23,RPL11,RPS2,RPS3和HSPA5(图4f)。研究数据揭示营养饥饿减缓了RPL27a与METTL3的相互作用,增加了RPL11和RPL23与METTL3的相互作用(图4f-g)。而且RPS27a,RPL23和RPL11的表达没有受到饥饿的显著影响(图4h)。这些结果说明这些蛋白质与METTL3的相互作用将影响METTL3蛋白的泛素化和稳定性。作者检测了MEFs中METTL3的表达来进一步验证上述结果,这些MEF转染了分别靶向RPS27a,RPL23和RPL11的siRNA,发现siRPS27a MEFs中METTL3上调,但RPL11或RPL23敲低的MEFs中METTL3没有显著变化(图4i)。因为RPS27a是一种泛素融合蛋白,它可以释放活性泛素单体,介导蛋白的泛素降解,所以作者研究了RPS27a是否导致METTL3泛素化。结果显示RPS27a强烈减弱的METTL3泛素化抑制(图4j)。这些结果表明,营养饥饿时RPS27a-METTL3相互作用受损导致METTL3泛素化抑制,从而增强METTL3稳定性。
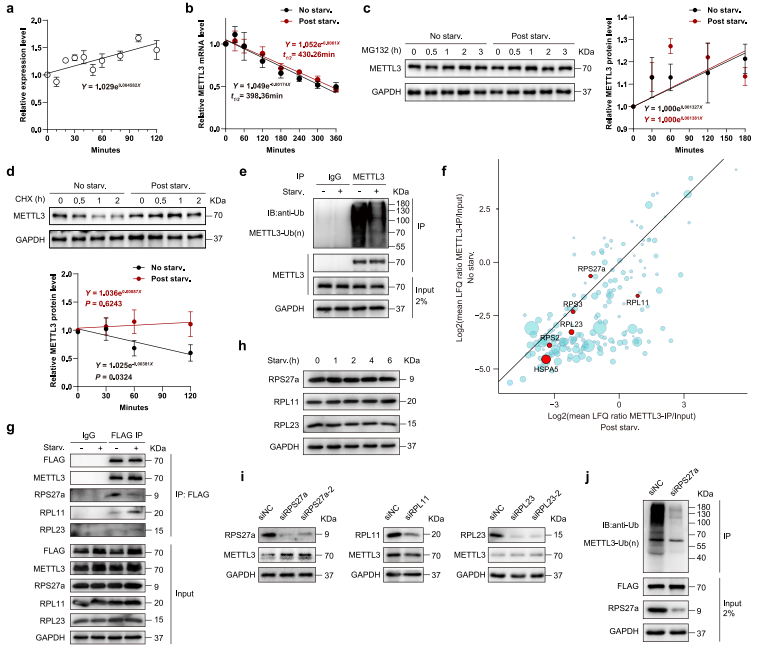
图4 受抑制的RPS27a-METTL3相互作用使METTL3在饥饿下稳定
5、YTHDF3识别饥饿诱导的FOXO3 mRNA的m6A高甲基化
接下来,作者尝试识别营养缺乏时与不同的YTHDF3结合的mRNA。发现了1041个上调的,1814个下调的转录因子(图5a)。分析这些结合位点,m6A核心基序“GGAC”被高度检测到(图5b)。大多数的这些结合位点定位在蛋白编码转录和高度聚集在CDS和3’UTR区域,特别是在停止密码子附近(图5c),与m6A峰的分布模式一致。因此,作者推断在营养缺乏时mRNA能与YTHDF3结合主要依赖于m6A修饰。
随后,作者发现与正常组织相比,营养缺乏时期,共2811个mRNA的m6A峰增多和2552个mRNA的m6A峰减少(图5d)。
为鉴别营养缺乏时与YTHDF3结合增加潜在的m6A高甲基化靶点,作者将营养缺乏时YTHDF3 RIP-seq富集峰与超m6A峰交叉,得到了86个峰(图5e)。在这86个峰中,有7个基因被注释到GO-term自噬(0006914),包括FOXO3,BMF,DDIT3,AP4M1,SESN2,ZFYVE1和ZFYVE26(图5e)。RIP-qPCR实验结果表明,YTHDF3营养缺乏时,这些自噬相关转录本中FOXO3富集的最为显著(图5i),并且都依赖于METTL3。
根据研究数据显示,当营养缺乏时,FOXO3 mRNA中的m6A高甲基化峰位于终止密码子周围的CDS和3'UTR区域(图5f)。MeRIP-qPCR分析验证沉默METTL3减少了由于营养缺乏而在FOXO3转录本停止密码子周围CDS和3'UTR区域的m6A峰,也减弱了m6A高甲基化水平(图5g-h)。与FOXO3 mRNA衰减的m6A同步,YTHDF3与FOXO3 mRNA的结合相应减少(图5i)。作者使用重组YTHDF3蛋白和含有不同FOXO3的CDS或序列的生物素化RNA探针进行了电泳迁移率转移试验(EMSAs)3'UTR加或不加m6A修饰。结果表明,一旦从RNA探针上去除m6A修饰,无论m6A位于FOXO3的CDS或3'UTR中,YTHDF3与探针之间的相互作用都显著减弱(图5j-k)。此外,由于与YTHDF3结合的探针1或3的观察到的带强度显著高于与YTHDF3结合的其他探针,作者推断FOXO3-CDS上2158 nt、2151 nt、2163 nt(探针1)和2295 nt(探针3)的m6A位点对于YTHDF3识别FOXO3终止密码子区域可能比其他m6A位点更关键(图5j-k)。总之,这些结果表明,在营养缺乏期间,METTL3介导的m6A高甲基化是YTHDF3 - FOXO3 mRNA相互作用所必需的。
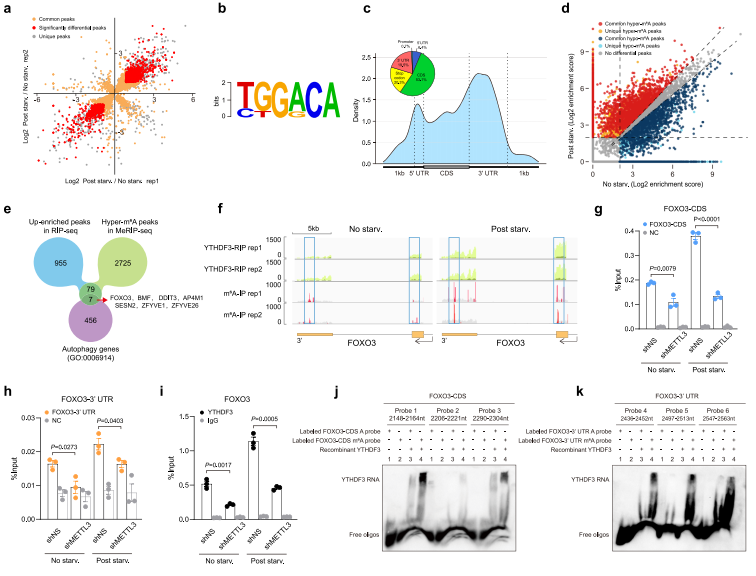
图5 YTHDF3识别饥饿诱导的FOXO3 mRNA的m6A高甲基化
6、FOXO3是YTHDF3促进自噬的关键
为研究YTHDF3在调节FOXO3表达中的作用,研究敲低YTHDF3,发现在正常和营养饥饿条件下,FOXO3蛋白水平都显著降低(图6a)。相应地,YTHDF3过表达导致FOXO3表达增加(图6a)。沉默YTHDF3导致参与自噬起始、成核、扩张和自噬溶酶体融合的FOXO3靶点的mRNA水平降低,而过表达YTHDF3导致这些基因的表达上调(图6b)。WB实验进一步验证了这些变化(图6c)。数据显示,在营养匮乏时,p-FOXO3(S413)水平在细胞核中增加,在细胞质中减少,其方式与总组分类似(图6a)。另一方面,作者检测到YTHDF3 KD细胞中pFOXO3(S413)水平下降,而YTHDF3过表达导致相反的效果。然而,磷酸化FOXO3与泛FOXO3的比值不受明显影响(图6a)。这些结果表明,YTHDF3可能在调节FOXO3的翻译中起关键作用,而不是FOXO3翻译后修饰如磷酸化。通过沉默或过表达YTHDF3,大部分FOXO3参与自噬的靶基因包括在mRNA和蛋白水平上均有显著变化。然而,四个基因(BECN1、ATG12、ATG4A和ATG4C)的蛋白水平变化非常轻微,而它们的转录水平却发生了显著变化(图6b-c)。作者认为这可能是由于基因特异性和细胞类型依赖性基因表达规则的差异。接下来,作者研究了METTL3缺失后FOXO3的表达情况。在正常和饥饿条件下,敲低METTL3明显抑制了YTHDF3过表达MEFs细胞核和细胞质中FOXO3的表达水平(图6d)。在METTL3沉默的细胞中,细胞核中的FOXO3明显低于对照细胞,因为它在饥饿的反应中转移到细胞核(图6d)。此外,参与自噬的FOXO3靶基因的大部分蛋白水平也被METTL3敲除减弱,而被METTL3过表达增强(图6e)。然而,在沉默或过表达METTL3的MEFs中也检测到FOXO3靶点表达的差异(图6e)。综上所述,这些数据进一步表明YTHDF3具有调节作用FOXO3的表达和改变FOXO3靶向自噬基因的表达依赖于METTL3。
为确定受损的FOXO3表达是否导致YTHDF3缺陷细胞的自噬功能障碍,研究进行了拯救实验(图6g)。在YTHDF3缺陷的细胞中异位表达FOXO3,挽救了在Baf.A1存在和不存在的情况下自噬标志物水平的降低,挽救了p62的降解(图6h),恢复了FOXO3靶基因的表达水平(图6i-j)。利用mCherry-GFP-LC3报告载体,作者证明了FOXO3的异位表达极大地恢复了YTHDF3缺陷引起的自噬体形成和自噬流缺陷(图6k-l)。为进一步确定FOXO3在YTHDF3调节自噬中的作用,敲除FOXO3并检测YTHDF3的实验数据显示,沉默FOXO3显著阻碍YTHDF3促进的LC3-II水平上调和p62降解(图6m)。mCherry-GFP-LC3报告基因实验表明,敲除FOXO3抑制饥饿时YTHDF3过表达诱导的自噬体和自溶酶体的形成(图6n-o)。这些结果表明FOXO3是YTHDF3促进自噬的关键功能靶点。

图6 FOXO3是YTHDF3促进自噬的关键
7、YTHDF3促进FOXO3翻译,但是不影响其mRNA的稳定性
作者想知道YTHDF3调控FOXO3表达的机制。敲除YTHDF3并没有影响FOXO3 mRNA的水平(图7a),这表明YTHDF3可能对FOXO3的翻译有影响。多聚体分析证明了这种破坏YTHDF3在40S/60S核糖体和80S单体部分显著增加,在多聚体部分显著减少(图7b)。YTHDF3缺陷细胞的翻译池(多体)中FOXO3 mRNA的水平低于对照细胞(图7c),表明YTHDF3的缺失减弱了FOXO3的翻译。
为解决FOXO3-CDS中m6A位点对YTHDF3促进FOXO3转译的影响,作者生成了野生型和突变型FOXO3-CDS表达结构。上游CDS的m6A突变基序(FOXO3-CDS-mut2)作为对比。数据显示FOXO3-CDS-mut1而不是FOXO3- CDS -mut2消除了YTHDF3增强的FOXO3表达(图7d),这表明FOXO3- CDS中终止密码子附近的m6A位点在YTHDF3促进FOXO3的翻译中起着关键作用。荧光素酶报告基因检测YTHDF3促进FOXO3翻译。鉴定出的FOXO3的3’ UTR部分,被克隆到pEZX-MT06载体中报告基因萤火虫荧光素酶的3 ' UTR区域(图7e)。这些结果表明FOXO3-中终止密码子附近的m6A位点3'UTR区域也参与调节YTHDF3 FOXO3翻译。
此外,作者还研究了YTHDF3是否调节FOXO3 mRNA的稳定性。用转录抑制剂放线菌素D(actinomycin D,Act D)处理,在对照组和YTHDF3缺陷细胞组之间无显著差异(图7h),表明YTHDF3不影响FOXO3 mRNA的稳定性。数据表明,YTHDF3促进FOXO3的翻译取决于它对FOXO3 CDS和3'UTR区域停止密码子周围m6A位点的识别,但YTHDF3不影响FOXO3 mRNA的稳定性。
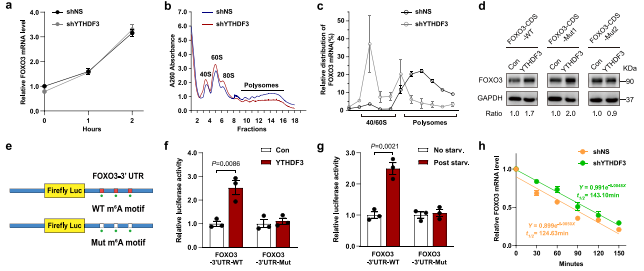
图7 YTHDF3促进FOXO3翻译,但是不影响其mRNA的稳定性
8、YTHDF3可能与eIF3a和eIF4B相互作用促进FOXO3的翻译
从免疫共沉淀(co-IP)LC-MS / MS数据中,作者注意到YTHDF3与多个翻译起始因子共纯化。eIF3a和eIF4B是营养缺乏时最显著上调的蛋白质(图8a-b)。对不同核糖体组分的WB分析发现,当YTHDF3缺失时,eIF3a和eIF4B从较重的多聚体组分转移到较轻的多聚体组分(图8c),表明eIF3a和eIF4B可能在YTHDF3对翻译的影响中发挥作用。silico docking分析显示eIF3a、eIF4B和YTHDF3之间存在潜在的蛋白质相互作用。
在对接能最低的对接模型(YTHDF3-eIF3a: - 1002.8; YTHDF3-eIF4B: - 834.7)中,以下残基被定位在模型界面并负责相互作用:YTHDF3中的Y424、E426和T441以及eIF3a中的R476、R483和I484;YTHDF3中的Y424、T441和S470以及eIF4B中的S88、F99和Y141(图8D)。co - IP实验发现在正常和无营养条件下,内源性eIF3a和eIF4B与YTHDF3共沉淀;营养饥饿可以同时增加沉淀的eIF3a、eIF4B和YTHDF3的数量。在YTHDF3 KD MEFs中,有少量蛋白沉淀(图8e)。接下来,作者在裂解液中添加RNase A并没有减少与YTHDF3分离的eIF3a和eIF4B的量,这表明这种相互作用是不依赖于RNA的(图8f)。为确定eIF3a和eIF4B对FOXO3翻译的影响,作者使用shRNA慢病毒敲低eIF3a或eIF4B(图8g)。敲低这两种蛋白中的任何一种都降低了FOXO3蛋白水平(图8g),表明FOXO3的翻译被削弱了。这些结果表明YTHDF3可能与eIF3a和eIF4B相互作用,促进FOXO3的翻译。Co-IP实验也证实了YTHDF3与PABP1或eIF4G2在MEFs中的相互作用。然而,这些相互作用在营养饥饿后显著减少(图8h),而YTHDF3和eIF3a或eIF4B之间的相互作用增加(图8e),表明YTHDF3和翻译起始调节因子之间的相互作用在不同的细胞亚型和不同的细胞应激条件下可能有很大的不同。
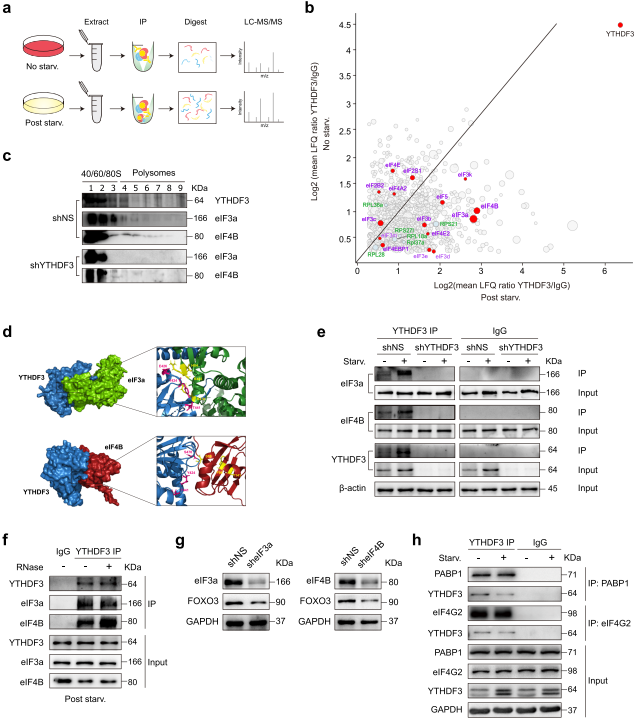
图8 YTHDF3可能与eIF3a和eIF4B相互作用促进FOXO3的翻译
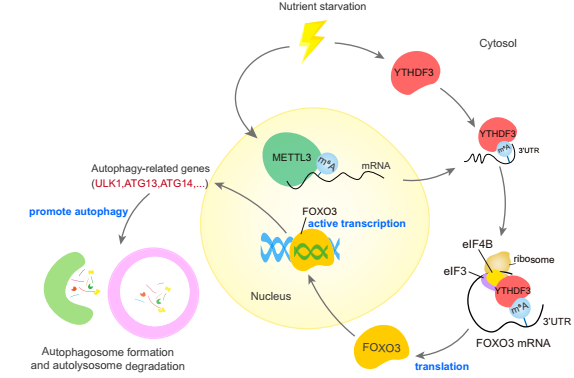
图9 机制图
结论
总的来说,本研究揭示了转录组学和自噬之间的重要联系。作者提供了m6A阅读器的证据YTHDF3作为一个营养应答器,结合m6A高甲基化,由METTL3安装在FOXO3 mRNA的停止密码子周围,然后YTHDF3募集eIF快速促进FOXO3的翻译,进一步在转录上激活核心自噬基因的一部分,从而促进自噬。
参考文献
Hao W, Dian M, Zhou Y, Zhong Q, Pang W, Li Z, Zhao Y, Ma J, Lin X, Luo R, Li Y, Jia J, Shen H, Huang S, Dai G, Wang J, Sun Y, Xiao D. Autophagy induction promoted by m6A reader YTHDF3 through translation upregulation of FOXO3 mRNA. Nat Commun. 2022 Oct 4;13(1):5845. doi: 10.1038/s41467-022-32963-0.