






METTL3通过抑制内质网应激增强宫颈癌的进展
m6A是mRNA上最丰富的化学修饰,在许多生物过程中起着重要作用。然而,m6A在宫颈癌(CC)肿瘤发生中的作用尚不清楚。在这里,我们发现METTL3, m6A甲基转移酶家族的核心成员,在CC中大幅上调。机制上,转录因子ETS1招募P300和WDR5分别介导METTL3启动子中H3K27ac和H3K4me3组蛋白修饰,诱导METTL3转录激活。此外,我们通过MeRIP-seq确定了TXNDC5为METTL3介导的m6A修饰的靶点,并揭示了METTL3介导的TXNDC5表达依赖于m6A阅读器依赖的方式。在功能上,我们通过体内外实验验证了METTL3通过调节TXNDC5的表达促进CC细胞增殖和转移。此外,我们的研究还验证了METTL3/ TXNDC5轴对内质网应激的影响。综上所述,METTL3促进了CC的恶性进展,提示METTL3可能是CC潜在的预后生物标志物和治疗靶点。本文于2022年8月发表于“Oncogene”(IF=8.756)上。
技术路线

结果
1)METTL3在CC中表达上调并与预后相关
METTL3通常在大多数人类癌症中起致癌基因的作用。为了研究METTL3在CC进展中的潜在作用,我们首先通过免疫组化染色确定了METTL3的表达。根据染色结果,正常组织中METTL3的表达水平低于CC组织(图1A, B)。CC组织中IF染色结果显示癌组织中METTL3的表达高于癌症前期组织(图1D)。为了检测宫颈上皮内瘤变(CIN)和CC组织中m6A的修饰水平,我们进行了IF、斑点杂交和m6A定量测定。结果显示,与正常组织相比,CIN或CC组织中m6A含量显著增加(图1E-G)。METTL3高表达的CC患者总生存率明显降低(OS,图1C)。OS的单因素和多因素Cox回归分析显示,METTL3是CC患者的独立预后因素(补充表,未展示)。以上数据证实,METTL3在CC组织中上调,与不良预后相关。

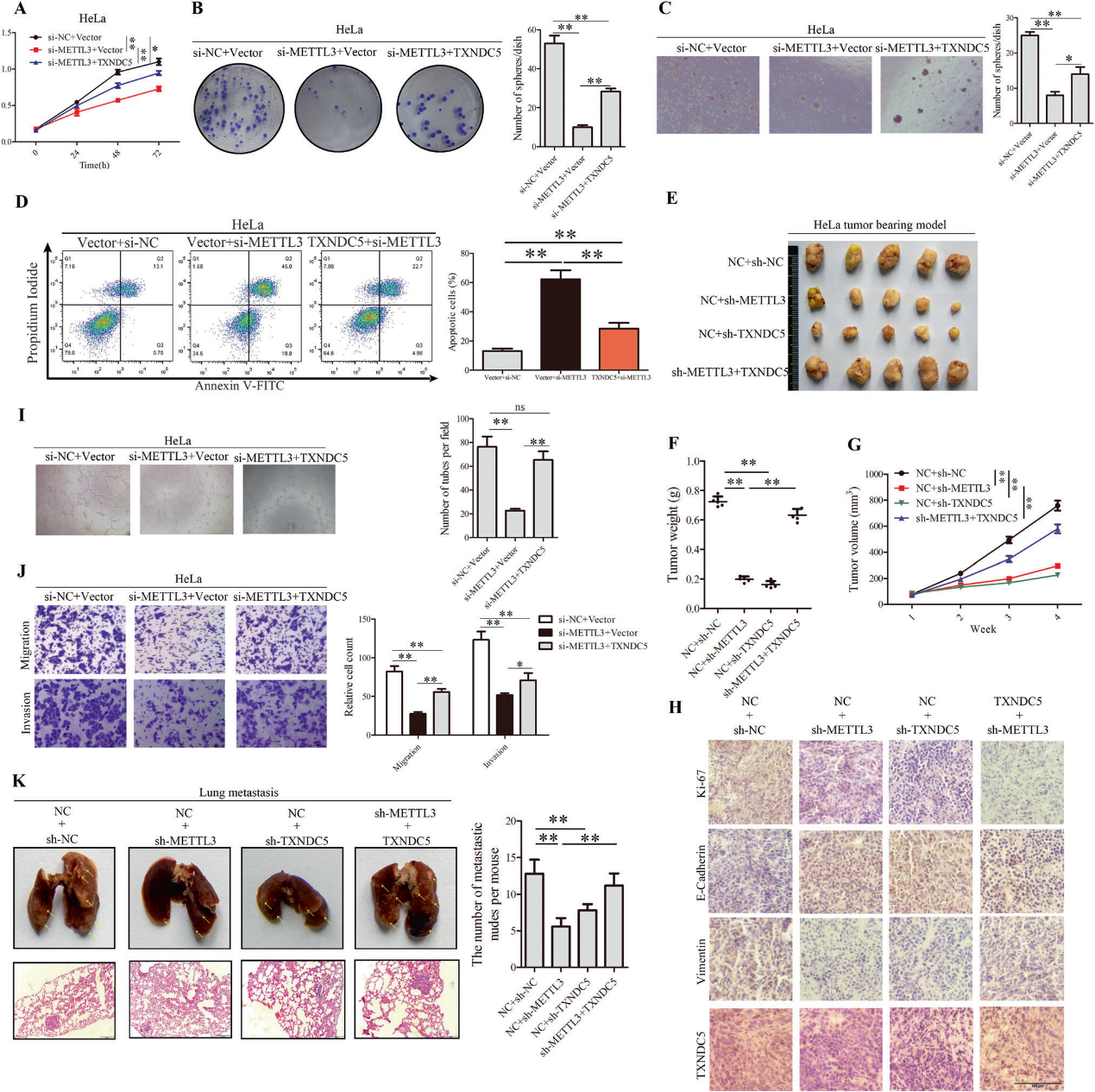
2)METTL3促进CC细胞在体外和体内的增殖和转移
为了弄清METTL3在CC进展中的作用,我们在HeLa和SiHa细胞中过表达并沉默METTL3。过表达METTL3显著促进细胞生长(图2A,B)。细胞凋亡检测结果表明,METTL3及其m6A催化活性抑制了细胞凋亡能力(图2C)。为了验证METTL3在体内的作用,我们还构建了肿瘤移植模型。统计结果显示,METTL3明显促进肿瘤生长(图2E-G)。为了了解METTL3对CC转移的影响,我们在体外进行了transwell试验。我们的数据表明,METTL3促进了CC细胞的迁移和侵袭(图2I)。为了进一步研究METTL3在CC恶性进展中的作用,我们进行了管形成实验,发现METTL3在CC细胞中有更强的作用(图2J)。Phalloidin染色显示,METTL3过表达后CC细胞中丝状肌动蛋白重组的侵袭性伪足发生了显著变化(图2H),揭示了METTL3在CC细胞运动中的重要作用。接着,我们进一步阐明了METTL3在体内CC转移中的作用。通过裸鼠尾静脉注射稳定转染METTL3和相应对照的HeLa细胞。4周后,METTL3过表达组肺转移灶增加(图2K, L),以上实验转染效率参照图2M。综上所述,METTL3在体内外均能促进CC细胞的增殖和转移。
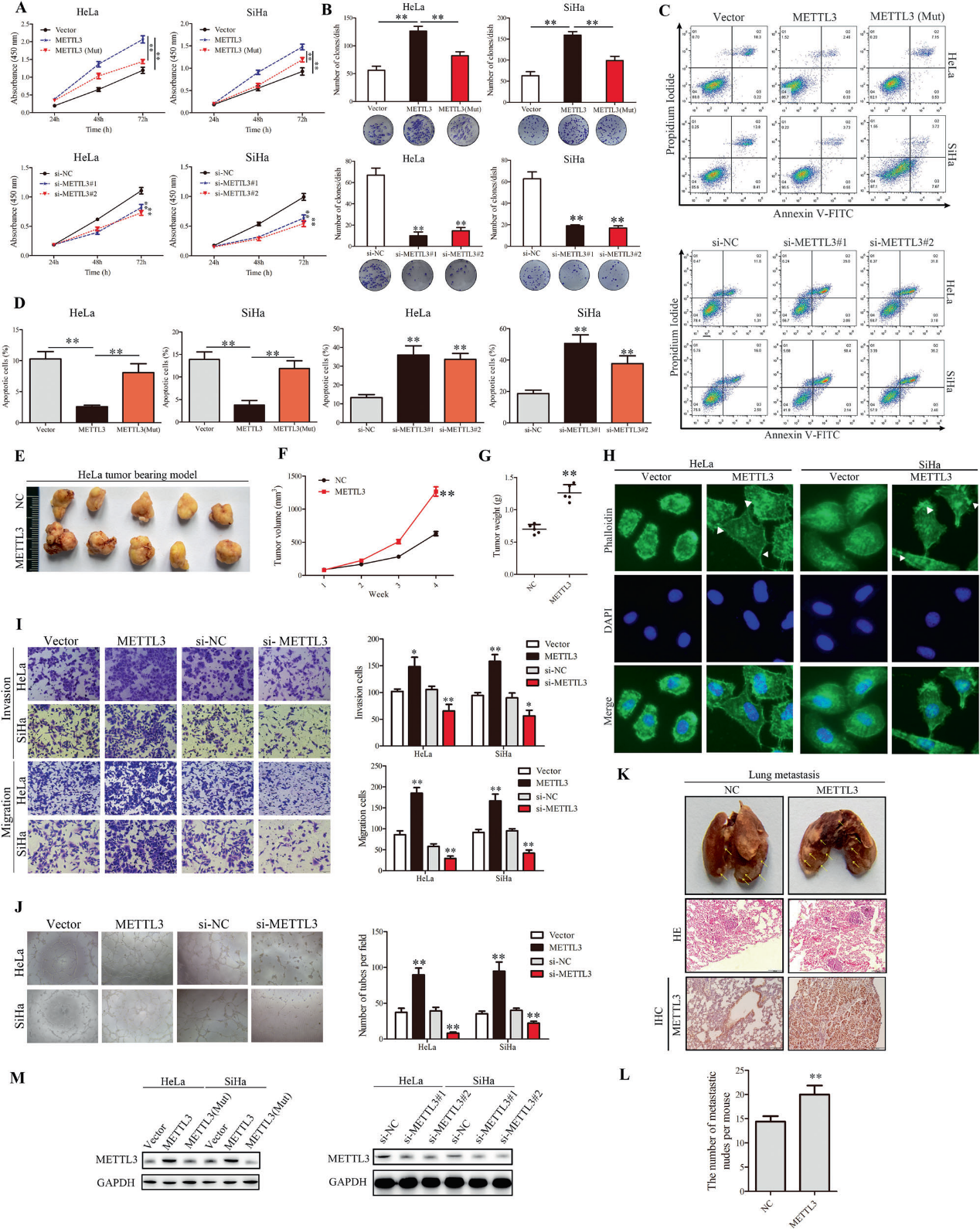
3)ETS1通过P300介导的H3K27ac和WDR5介导的H3K4me3转录激活METTL3启动子区
为了确定METTL3在CC中高表达的原因,我们通过UCSC基因组生物信息学网站分析了METTL3的启动子。在METTL3启动子区域发现了大量 H3K27ac和H3K4me3信号(补充图)。因此,我们进行了染色质免疫沉淀(ChIP)试验,验证了H3K27ac和H3K4me3信号确实在METTL3的启动子区富集(图3A)。敲低P300和WDR5后,METTL3的蛋白水平显著降低(图3C)。Western blot分析显示,C646和OICR-9429分别作为P300和WDR5的抑制剂处理后,METTL3蛋白减少,而NaBu作为组蛋白去乙酰化酶抑制剂处理后,METTL3蛋白增加(图3B)。荧光素酶报告基因分析表明,P300和WDR5的敲除明显削弱了荧光素酶的活性(图3D)。这些观察结果表明,P300介导的H3K27ac和WDR5介导的H3K4me3组蛋白修饰可能上调了METTL3。为了进一步研究METTL3在CC中上调的分子机制,我们结合不同转录因子预测网站的结果,在候选转录因子中, ETS1与CC的恶性进展相关,因此我们推测ETS1可能是METTL3的潜在转录因子。western blot结果显示,METTL3、H3K4me3和H3K27ac的蛋白水平与ETS1呈正相关,而组蛋白H3的蛋白水平保持不变(图3E, F)。RT-qPCR结果与western blot结果一致(图3G)。我们推测ETS1招募了WDR5和P300,并与METTL3启动子结合,介导其H3K4me3和H3K27ac组蛋白修饰,从而发挥转录激活作用。为了证实我们的猜想,我们确定了METTL3启动子的结合基序和5个结合位点。ChIP分析验证了ETS1与METTL3启动子结合的5个位点(图3I, J)。我们还构建了含有结合位点突变的METTL3启动子区域的荧光素酶报告基因质粒,得到了相同的结果(图3H)。IP检测显示ETS1和WDR5/ P300之间存在物理相互作用(图3K)。为了验证ETS1是否通过招募P300和WDR5来调节METTL3组蛋白修饰,我们进行了RT-qPCR,发现沉默P300或WDR5减弱ETS1诱导的METTL3 mRNA水平(图3L)。此外,ChIP分析结果显示,沉默P300和WDR5降低了ETS1诱导的METTL3启动子区H3K27ac和H3K4me3信号的富集(图3M, N)。以上数据证实ETS1可能通过招募介导METTL3启动子中H3K27ac和H3K4me3激活的P300和WDR5来上调METTL3。
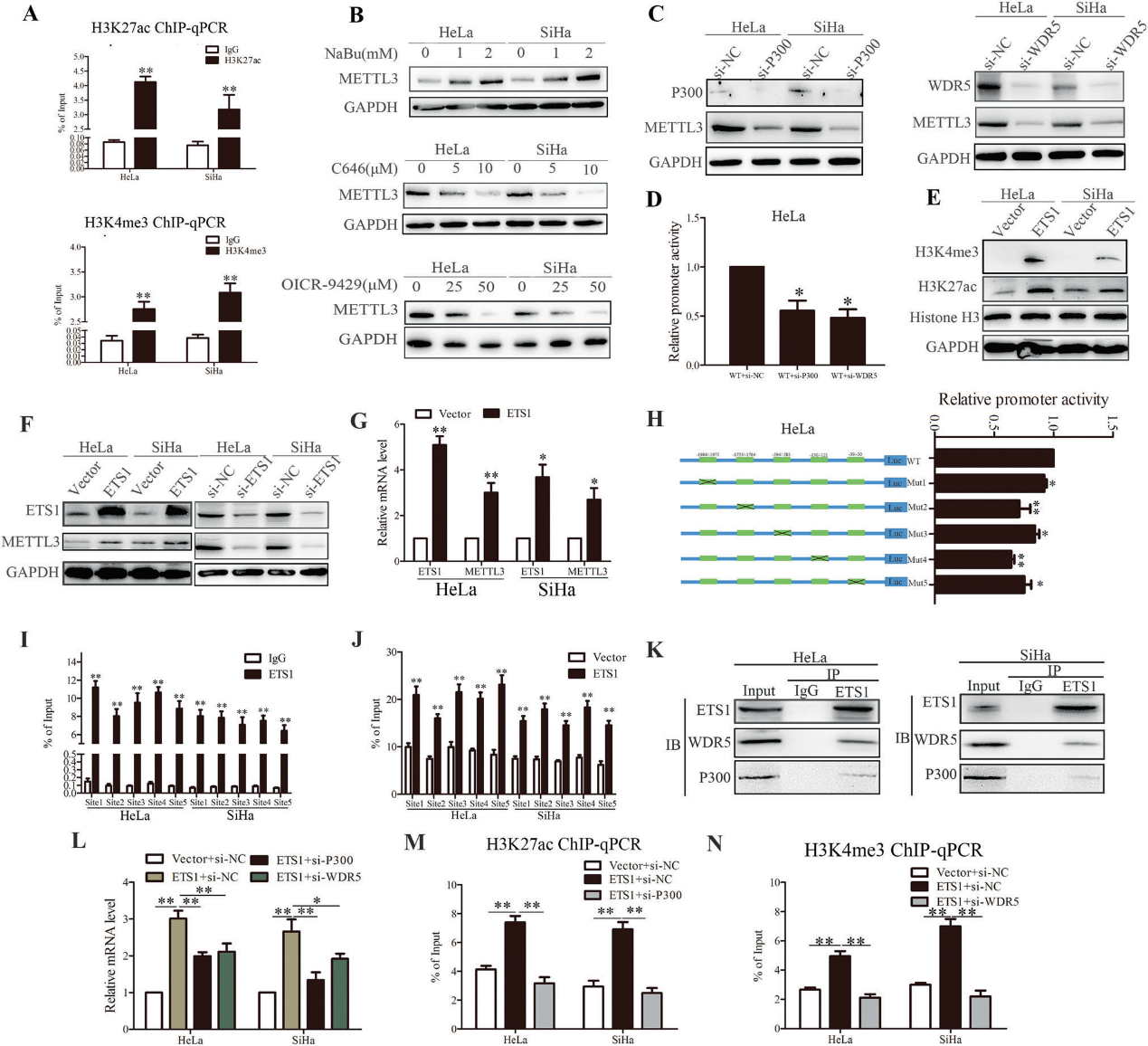
4)METTL3诱导的m6A修饰是TXNDC5上调的原因
为了进一步探讨CC恶性进展中与METTL3相关的分子机制,对转染METTL3和Vector质粒的HeLa细胞进行MeRIP-seq。m6A峰分布比例显示,m6A峰主要分布在3'UTR、5'UTR、CDS和终止密码子上(图4A)。KEGG通路分析显示CC中存在潜在的通路(图4B,C)。我们发现METTL3过表达后,ER中的蛋白处理发生了很大的变化。有趣的是,在RNA-seq和MeRIP-seq数据中重叠前10的转录本中,TXNDC5是一种ER蛋白,它有助于蛋白质的正确折叠(图4D)。因此,我们认为TXNDC5是METTL3的靶点。然后我们通过western blot和RT-qPCR验证了TXNDC5与METTL3在蛋白质和RNA水平上的正相关性(图4F-H)。同时,我们发现催化突变体METTL3对TXNDC5的上调比野生型要弱(图4G, H)。突变效率通过斑点杂交试验得到验证(图4E)。使用通用甲基化抑制剂DAA和m6A甲基化抑制剂Cyc阻断CC细胞的甲基化,我们发现TXNDC5在蛋白质和mRNA水平上均降低(图4I, J)。MeRIP-qPCR显示,在METTL3过表达后,TXNDC5的m6A水平显著升高(图4K)。总之,我们的数据证实了METTL3通过TXNDC5 mRNA的m6A甲基化促进TXNDC5。
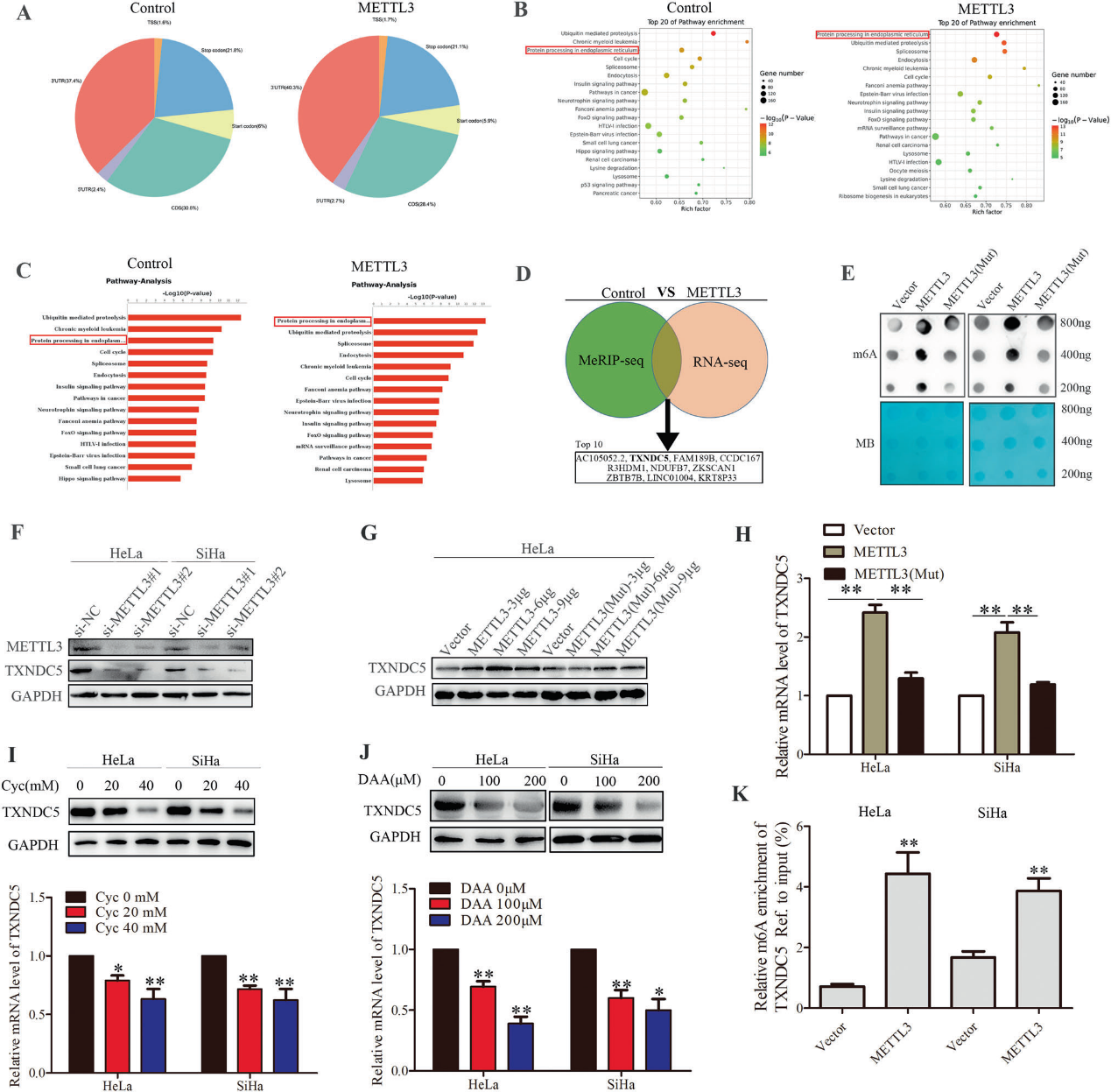
5)METTL3通过m6a -reader依赖通路促进TXNDC5的表达
我们想知道m6A阅读器是否参与了CC细胞中TXNDC5转录水平的变化。首先,我们通过western blot和RT-qPCR检测YTHDFs和IGF2BPs对TXNDC5表达的影响(图5B-D),发现YTHDF1/2和IGF2BP2/3在TXNDC5的调控中起重要作用。RIP-qPCR结果提示YTHDF1/2和IGF2BP2/3可与TXNDC5 mRNA相互作用。这些数据揭示了YTHDF1/2和IGF2BP2/3在TXNDC5 mRNA上的潜在靶向作用。接下来,我们探讨了m6A阅读器对TXNDC5的调控机制。由于敲除YTHDF1后,TXNDC5 mRNA保持不变,蛋白表达降低,证明YTHDF1可能在转录后水平调控TXNDC5。为了验证这一点,我们用蛋白质翻译抑制剂环己酰亚胺(CHX)处理METTL3稳定干扰和相应对照的HeLa细胞。结果表明,METTL3敲减与对照HeLa细胞之间TXNDC5蛋白的半衰期相似(图5A),因此m6A甲基化与TXNDC5蛋白的稳定性无关。因为翻译调节是YTHDF1的重要功能之一。由于eIF3a/3b可通过与YTHDF1相互作用调节mRNA的翻译,我们研究了eIF3a/3b对TXNDC5的影响。我们通过western blot排除了eIF3a/b对TXNDC5的影响(补充图)。综上所述,YTHDF1可能通过依赖m6A的方式促进TXNDC5 mRNA的翻译。有报道称m6A阅读器可以调节mRNA的稳定性,因此我们在CC细胞中检测了METTL3过表达或m6A阅读器抑制时TXNDC5 mRNA的稳定性。我们发现,敲低IGF2BP2/3会降低TXNDC5 mRNA的稳定性,而敲低YTHDF2则会增加TXNDC5 mRNA的稳定性。这些结果表明,IGF2BP2/3增强了TXNDC5 mRNA的稳定性,而YTHDF2可能促进TXNDC5 mRNA的降解(图5E)。同时,敲除这些阅读器可以挽救METTL3对TXNDC5半衰期、mRNA和蛋白水平的影响(图5F, H)。RIP-qPCR证实了阅读器与TXNDC5 mRNA之间的相互作用(图5G)。我们还研究了METTL3对YTHDF2表达的影响(图5I),这表明METTL3可能通过下调YTHDF2抑制CC中TXNDC5 mRNA的降解。综上所述,说明METTL3通过m6A-reader依赖的方式调控TXNDC5的表达。
6)沉默的METTL3逆转TXNDC5对CC细胞增殖和转移的影响
我们在有无si-METTL3的情况下过表达TXNDC5,发现TXNDC5有挽救作用(图6A-C, I, J),并且TXNDC5降低了si-METTL3增加的凋亡率(图6D)。值得注意的是,除了血管生成实验被完全拯救外,所有其他表型均显示TXNDC5对si-METTL3的部分拯救。此外,TXNDC5的过表达逆转了sh-METTL3对肿瘤生长的下调(图6E-G)。METTL3和TXNDC5的下调可减少CC细胞的肺转移。TXNDC5的过表达挽救了sh-METTL3对肺转移的下调(图6K)。IHC显示sh-METTL3导致异种移植瘤组织中E-Cadherin水平升高,Ki-67、Vimentin和TXNDC5水平降低(图6H)。总的来说,METTL3通过上调TXNDC5的表达促进CC的增殖和转移。
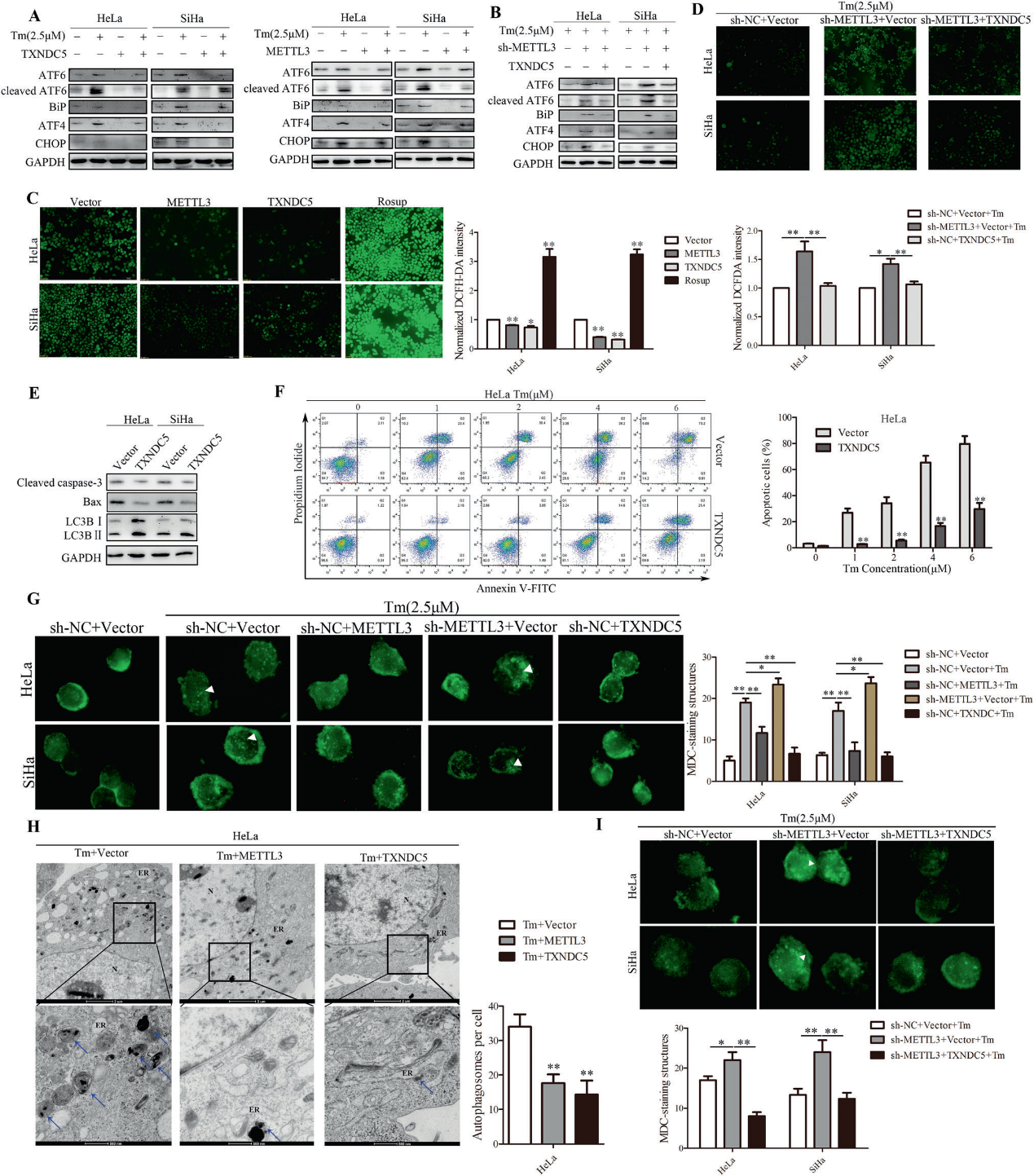
7)METTL3通过TXNDC5减少内质网应激引起的CC细胞死亡
为了研究METTL3/TXNDC5对CC内质网应激的影响,TXNDC5在经tunicamycin (Tm)处理或未处理的CC细胞中过表达。western blot结果显示,METTL3和TXNDC5抑制了Tm处理的内质网应激标志物的上调(图7A)。此外,我们发现过表达METTL3或TXNDC5会降低ROS水平(图7C)。我们通过透射电镜观察到转染了METTL3和TXNDC5的CC细胞亚细胞结构中ER的肿胀减少了(图7H)。为了研究TXNDC5对内质网应激的影响是否依赖于METTL3,我们进行了western blot,发现TXNDC5部分减弱了Tm处理的sh-METTL3引起的内质网应激标志物的上调(图7B)。CCCP可作为ROS诱导剂。ROS检测显示TXNDC5挽救了Tm/CCCP处理下shMETTL3诱导的ROS水平(图7D)。此外,TXNDC5过表达减少细胞凋亡和自噬(图7E)。如图7F所示,过表达TXNDC5后内质网应激诱导的细胞凋亡明显减少。然后我们观察到,与对照细胞相比,在Tm处理下过表达METTL3或TXNDC5的CC细胞显示出MDC标记的囊泡数量显著减少,而sh-METTL3导致相反的结果(图7G)。TEM分析显示,METTL3和TXNDC5与自噬体减少相关(图7H)。这些结果表明,METTL3和TXNDC5均抑制内质网应激介导的细胞凋亡和自噬。TXNDC5逆转了sh-METTL3对Tm处理下MDC标记囊泡数量的影响(图7I)。这些数据表明,METTL3可能通过TXNDC5抑制内质网应激介导的细胞凋亡和自噬。
结论:
我们的研究结果证实了METTL3在CC恶性进展中的致癌作用。机制上,ETS1将P300和WDR5招募到METTL3启动子上,并通过维持H3K27ac和H3K4me3状态促进其转录激活。我们还发现TXNDC5是METTL3的靶点,并指出了m6A阅读器介导的机制。此外,METTL3通过上调TXNDC5促进CC细胞增殖和转移,抑制内质网应激诱导的细胞凋亡和自噬。ETS1/METTL3/TXNDC5/内质网应激轴可能为CC的治疗提供新的见解。
参考文献:
Du QY, Huo FC, Du WQ, Sun XL, Jiang X, Zhang LS, Pei DS. METTL3 potentiates progression of cervical cancer by suppressing ER stress via regulating m6A modification of TXNDC5 mRNA. Oncogene. 2022 Sep;41(39):4420-4432. doi: 10.1038/s41388-022-02435-2.