






乳酸-NAD+轴通过下调p62激活肿瘤相关成纤维细胞
p62水平降低与癌症相关成纤维细胞(CAF)表型的诱导相关,CAF表型通过炎症和代谢重编程在体内外促进肿瘤发生。然而,肿瘤细胞如何下调基质成纤维细胞中的p62来驱动CAF激活是该领域尚未解决的核心问题。近日,有研究发现肿瘤分泌的乳酸下调了间质成纤维细胞中的p62;乳酸降低NAD+水平和PARP-1活性,从而下调间质p62;AP-1对乳酸诱导的间质成纤维细胞p62损失至关重要;PARP-1抑制剂(奥拉帕利)通过p62促进间质成纤维细胞的结缔组织反应。该研究探究了癌细胞促进前致瘤性CAF表型的分子机制,为抑制CAF活性提供新的治疗策略。该研究于2022年5月发表在《CELL REPORTS》,IF:9.995。
技术路线:

主要研究结果:
1. 前列腺癌细胞分泌一种可溶性因子,可降低基质成纤维细胞中p62的表达
为了研究p62在肿瘤间质中表达下调的机制,作者建立了小鼠GFP标记的前列腺基质细胞(mPSCs)与TRAMPC2前列腺癌(PCa)上皮细胞混合的体外细胞系统。与肿瘤细胞孵育时,基质细胞中Sqstm1(编码p62) mRNA水平降低,这也与真正CAF标志物(如Tgfb1和Sdf-1)的增加相关(图1A)。为了验证p62的下调是否需要肿瘤细胞和基质细胞之间的细胞接触,将人前列腺基质成纤维细胞(WPMY-1)与不同的PCa细胞系在双腔环境中共培养。在这些条件下,基质细胞中p62的蛋白和mRNA水平也下调(图1B和图1C),表明由PCa上皮细胞分泌的可溶性因子足以下调基质细胞中的p62。来自PCa细胞的条件培养基(CM)孵育基质细胞有效降低了基质细胞中的p62蛋白和mRNA水平,而正常前列腺上皮细胞(PrEC或RWEP1)的CM没有任何影响(图1D和1E)。为了确定SQSTM1 mRNA的减少是否是由于转录活性的抑制,使用了SQSTM1启动子控制下的荧光素酶报告基因。PCa CM降低了荧光素酶报告的活性(图1F),表明基质p62的下调是通过肿瘤细胞分泌的可溶性因子抑制SQSTM1启动子介导的。为了鉴定这种假定的肿瘤来源的可溶性因子,对PC3细胞的CM进行了大小分级。在成纤维细胞中,未分割的肿瘤CM和<3-kDa片段均在蛋白和mRNA水平上下调p62(图1G和1H),以及SQSTM1启动子驱动的荧光素酶报告基因的活性(图1I)。然而,>3-kDa部分在这些试验中完全不活跃(图1G-1I)。活性可溶性因子具有热稳定性(图1J-1L),提示其可能为代谢物。
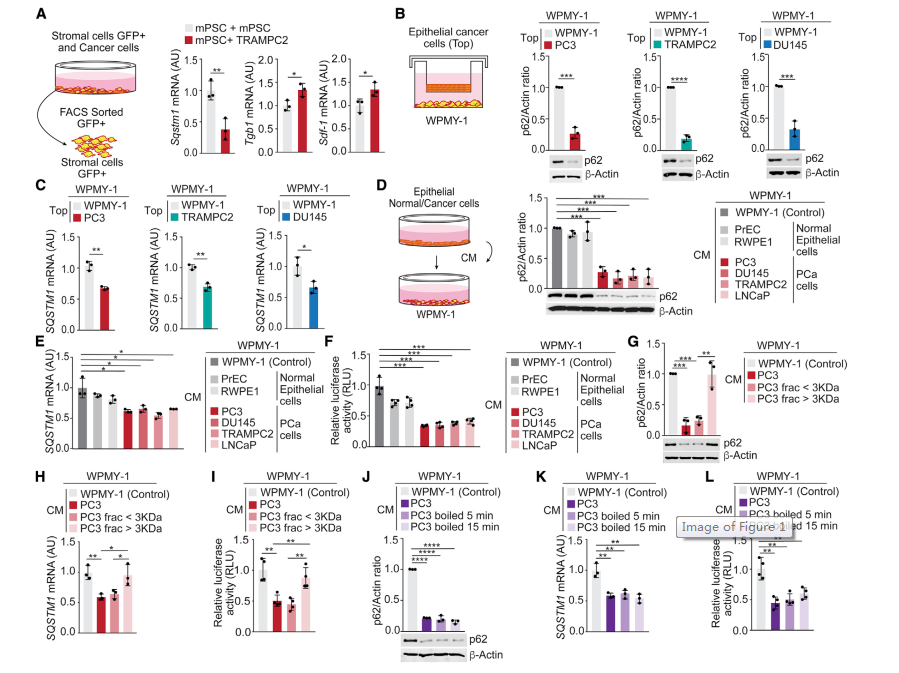
图1 PCa细胞分泌一种可溶性因子,可降低基质细胞中p62的表达
2. 乳酸下调间质成纤维细胞中的p62
由于癌细胞分泌大量的乳酸,并且由于Warburg效应在TME中积累,推测乳酸可能是导致p62下调的代谢物。正如预期的那样,PCa细胞系分泌的乳酸量高于正常前列腺上皮细胞(图2A)。由于乳酸的细胞摄取是由不同的单羧酸转运蛋白(MCT1、MCT2、MCT3和MCT4)介导的,接下来测试了阻断这些转运蛋白是否可以损害乳酸诱导的p62下调。MCT1在WPMY-1细胞中控制乳酸的细胞输入,敲低MCT1导致PC3细胞的CM诱导的p62下调受到抑制(图2B-2D)。同样,当PC3细胞中控制乳酸输出的MCT4被敲低时,WPMY-1细胞中p62的下调也被消除(图2E-2G和S1B)。MCT1抑制剂AZD3965处理PC3 CM处理的WPMY-1细胞后,p62下调受损(图2H-2J)。这些结果表明,PCa细胞分泌乳酸和基质细胞摄取乳酸是基质p62下调的关键步骤。为了证明乳酸足以下调成纤维细胞中的p62,将WPMY-1细胞与不同浓度的乳酸孵育24或48小时,这导致p62以时间和剂量依赖的方式下调(图2K-2N)。在基质细胞中下调p62所需的乳酸量(图2M和2N)与PCa细胞分泌的乳酸量(图2A)相似,也与体内肿瘤的乳酸量相似。然而,简单的培养基酸化不足以下调p62(图2O)。这些结果表明,上皮PCa细胞分泌的乳酸在转录水平上损害了基质细胞中p62的表达。
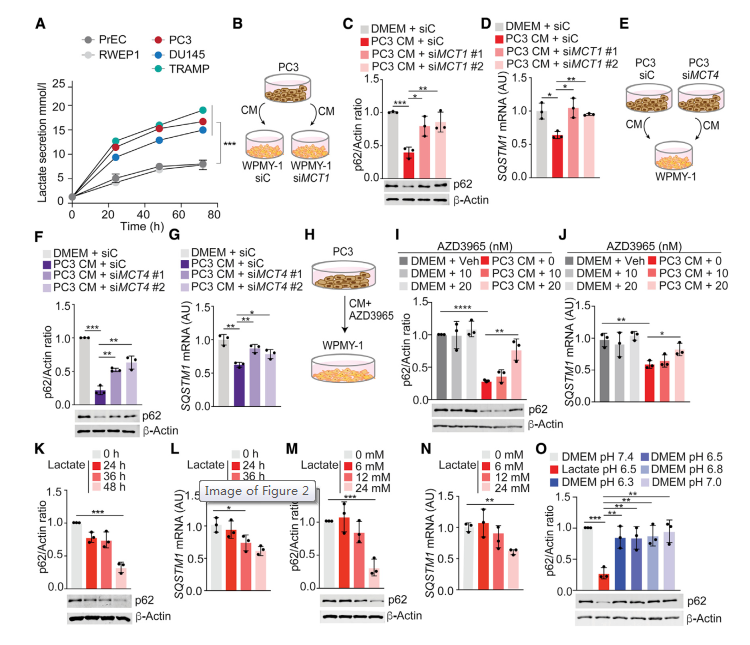
图2 乳酸下调p62
3. AP-1通过乳酸抑制p62下调
为了阐明乳酸下调基质p62的机制,下一步通过转座酶可及染色质测序全基因组分析(ATAC-seq)研究了乳酸处理的成纤维细胞的染色质可及性。这项分析显示,在乳酸处理的细胞中,染色质可及性普遍降低(图3A)。HOMER(基序富集的超几何优化)分析表明,在乳酸处理的细胞的封闭染色质区域中,AP-1转录因子显著富集(图3B)。对SQSTM1启动子的分析表明存在三个AP-1调节位点,这些位点也表明在乳酸处理的条件下染色质可及性降低(图3C)。与此一致,乳酸抑制了AP-1驱动的荧光素酶报告基因(图3D),这共同表明SQSTM1启动子中AP-1位点的抑制可以解释乳酸对基质p62表达的影响。
为了确定AP-1位点在SQSTM1启动子中的作用,分别突变了这些位点(AP-1A, AP-1B和AP-1C),并确定这些突变对SQSTM1启动子活性的影响。AP-1A的突变使SQSTM1启动子完全失活,达到与乳酸产生的水平相当的水平,而AP-1B或AP-1C的突变没有影响(图3E),这表明AP-1A是SQSTM1启动子中介导乳酸抑制的关键增强子元件。为了验证这一假设,通过CRISPR-Cas9编辑选择性地删除内源性SQSTM1启动子中的AP-1A元件,从而产生4种独立的WPMY-1细胞培养物(sgAP-1A),这导致p62 mRNA和蛋白表达受到抑制(图3F-3H)。这些结果确立了AP-1A在基质细胞中调节p62表达的相关性。与这一观点一致, ChIP分析表明,乳酸显著减少了c-FOS和c-JUN募集到AP-1A调节位点,但没有减少FOSB或JUNB的募集,且未检测到与AP-1B或AP-1C位点的结合发生变化(图3I)。下调c-FOS、c-JUN和JNK,或者用JNK的两种药理抑制剂抑制c-JUN磷酸化,在蛋白和mRNA水平上模拟了乳酸对p62表达的影响(图3J-3M)。这些数据表明,c-JUN/c-FOS AP-1转录复合物募集受损在乳酸诱导的基质p62下调中起关键作用。

图3 AP-1通过乳酸抑制p62的下调
4. 乳酸代谢降低NAD+水平介导p62下调
研究成纤维细胞中细胞外乳酸的命运。应用2H和13C同位素示踪和质谱方法来量化细胞外乳酸对NADH和中心碳代谢的贡献。细胞外乳酸通过LDH活性代谢,将氢原子贡献到NADH池并再生胞质烟酰胺腺嘌呤二核苷酸(NAD+)池(图4A和4B)。接下来,发现大约20%的[13C]乳酸示踪剂在三羧酸(TCA)循环中间产物上,这表明细胞外乳酸被代谢为促进线粒体TCA循环代谢的碳源(图4A和图4C)。测定在10 mM外源[13C]乳酸存在下的乳酸摄取和分泌通量的实验表明,WPMY-1细胞在分泌未标记的乳酸的同时摄取标记的乳酸(图4A和图4D)。这些结果与以下事实一致:乳酸可以可逆性地转化为丙酮酸盐,随后NAD+水平减少,有利于NADH的生成。增加培养基中丙酮酸的水平,使这一反应向相反的方向发生,显然损害了乳酸下调p62的能力(图4E和图4F)。因此,NAD+/NADH比率的变化可能有助于乳酸调节p62水平的作用机制。与这一假设一致,乳酸处理的基质成纤维细胞中的NAD+水平低于未处理的细胞(图4G)。使用NAD+或NAD+前体烟酰胺核苷(NR)治疗完全挽救了乳酸诱导的p62减少,且独立于自噬(图4H-4K)。
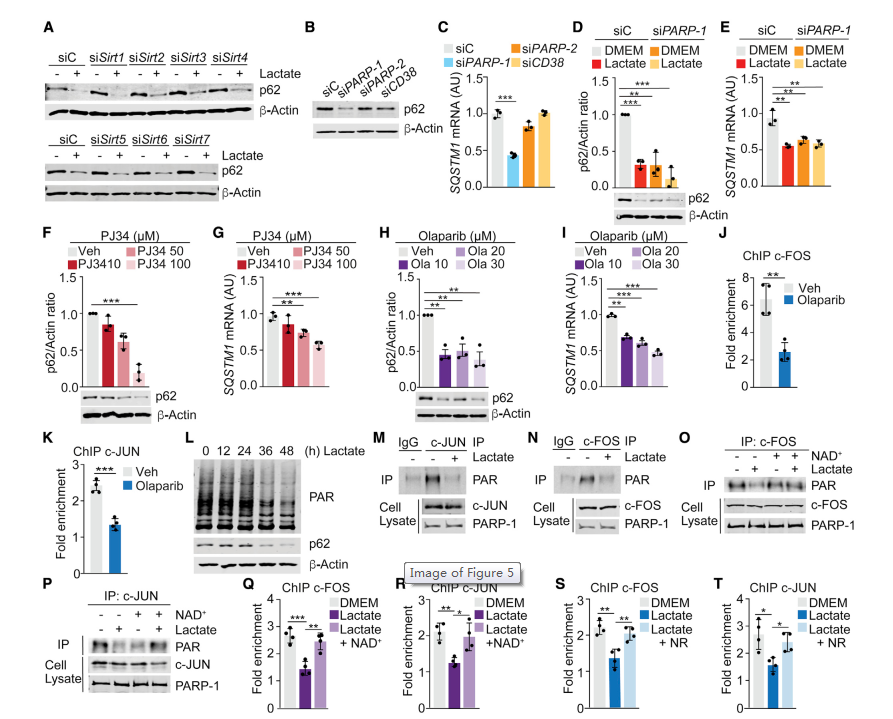
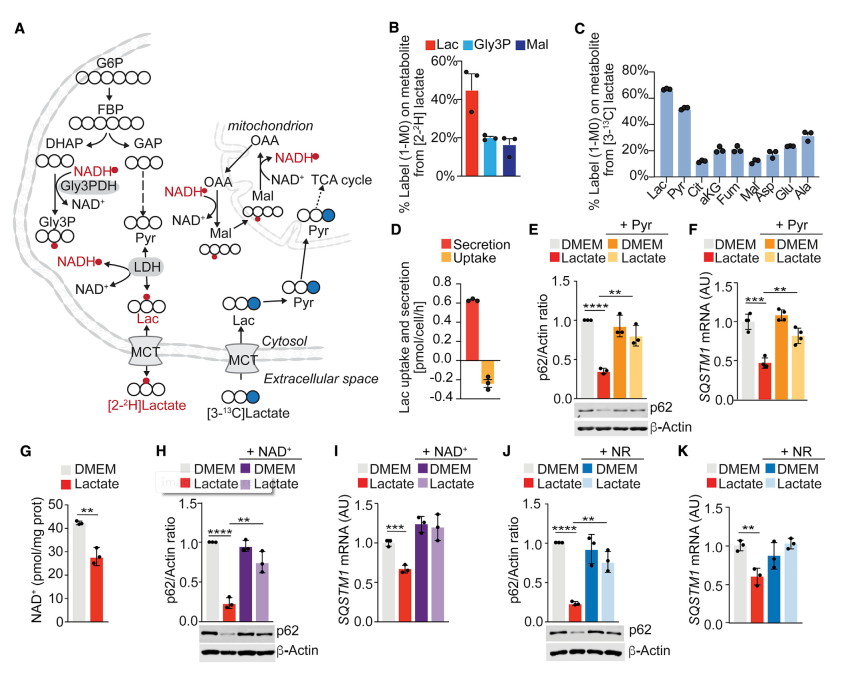
图4 乳酸改变NAD+/NADH水平介导p62下调
5. 乳酸抑制PARP-1可下调间质细胞p62
NAD+是三种转录和翻译后调节因子的辅因子,包括sirtuins (SIRT1到SIRT7)、cyclic ADP-核糖合酶(CD38和CD157)和PARPs (PARP-1/2)。其中,只有PARP-1的敲低或CRISPR-Cas9缺失完全模拟了乳酸对p62表达的影响(图5A -5E)。此外,PARP-1抑制剂PJ34或奥拉帕利也以剂量依赖性方式降低了p62的表达(图5F-5I)。PARP-1除了在DNA损伤修复(DNA damage repair, DDR)中发挥众所周知的作用外,还控制许多其他的生物过程,包括转录。此外,奥拉帕利抑制了c-FOS和c-JUN向SQSTM1启动子中AP-1A结合位点的募集(图5J和图5K)。这些结果表明,基质细胞中乳酸代谢触发的NAD+水平的变化对PARP-1活性的调节是p62下调的工具。因此,我们假设乳酸可以影响PARP-1的活化,从而影响c-JUN和c-FOS的聚腺苷二磷酸核糖基化。乳酸处理的细胞总poly(ADP-ribosyl)ation水平降低,c-JUN和c-FOS的poly(ADP-ribosyl)ation受损(图5L-5N)。在乳酸处理的细胞中添加NAD+可恢复这两种转录因子的poly(ADP-ribosyl)ation (图5O和5P),以及它们向SQSTM1启动子中AP-1调节位点的募集(图5Q和5R)。这些数据表明,乳酸代谢导致的NAD+耗损以及随后c-FOS和c-JUN的poly(ADP-ribosyl)ation受损是乳酸下调基质成纤维细胞中p62的机制基础。
图5 抑制PARP-1可下调间质细胞p62水平
6. AP-1在p62缺失驱动的CAF激活中至关重要
p62缺失可驱动基质细胞中的CAF表型,从而促进肿瘤进展。由于PCa细胞的乳酸分泌对于下调基质中的p62是必要和充分的,因此下一步确定乳酸是否驱动CAF表型。乳酸处理成纤维细胞增加了CAF活化的真正标志物的表达,如ACTA2和TGF-β,这些标志物的表达被添加NAD+逆转(图6A和6B)。对乳酸处理的细胞中差异表达基因的基因集富集分析(GSEA)显示,来自人类基质PCa数据集(GEO: GSE34312)的CAF signatures富集(图6C)。此外,SQSTM1启动子内源性失活的sgAP-1A成纤维细胞也显示出CAF标志物表达增加(图6D)。同样,在双腔共培养系统中,成纤维细胞sgAP-1A促进PC3细胞的迁移和侵袭,而sgC没有作用(图6E-6G)。PC3细胞与sgC和sgAP-1A成纤维细胞共培养表明,与sgC细胞相比,sgAP-1A细胞增强了PCa上皮细胞的侵袭和增殖指数(图6H和6I)。此外,与sgC成纤维细胞共植入相比,PC3细胞与sgAP-1A成纤维细胞共植入皮下异种移植导致肿瘤生长增加(图6J-6L)。由sgAP-1A成纤维细胞生成的肿瘤显示基质活化标志物增加,如胶原沉积、透明质酸(HA)和α-平滑肌肌动蛋白(α-SMA)(图6M)。这些结果证实SQSTM1启动子中的AP-1调节位点是乳酸驱动信号的靶点,并且在基质细胞内源性p62水平的控制中具有关键作用,对CAF表型的诱导至关重要。
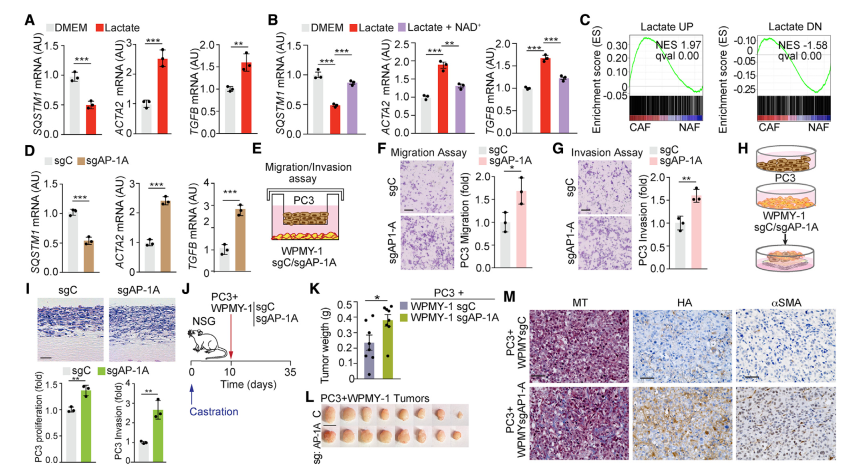
图6 AP-1控制基质细胞中SQSTM1启动子对PCa肿瘤的生长和侵袭至关重要
7. PARP-1抑制剂通过p62促进间质成纤维细胞的结缔组织反应
根据作者的模型,乳酸通过抑制PARP-1触发基质成纤维细胞中p62的下调。同样,通过CRISPR-Cas9敲除PARP-1模拟了乳酸的作用,并诱导了ACTA2和TGF-β等CAF活化标志物(图7A)。因此,假设PARP-1抑制剂(如奥拉帕利)足以诱导基质激活。奥拉帕利降低了SQSTM1 mRNA,并上调了ACTA2、SFRP1、TGF-β、MMP9和HA合酶等CAF标志物(图7B)。此外,对奥拉帕利处理的细胞进行的ATAC-seq分析表明,与乳酸处理相似,整个基因组中,在AP-1转录因子(包括SQSTM1启动子)的共有基序区域,染色质可及性显著降低(图7C)。PC3细胞与奥拉帕利预处理的WPMY-1细胞共培养导致PCa细胞在基础条件下的迁移和侵袭增加,这一情况在雄激素剥夺(ADT)下更为明显(图7D-7F)。此外,奥拉帕利处理的WPMY-1 CM导致PC3增殖增加(图7G和7H)。根据这些观察结果,假设奥拉帕利治疗后基质激活可能会抑制其抗肿瘤活性。为了检验这一假设,在基础或ADT条件下将GFP标记的PC3细胞(PC3GFP)与WPMY-1细胞(共培养)或不与WPMY-1细胞(单独培养)共培养,并且使用或不使用奥拉帕利治疗10日(图7I)。与单独培养条件下的处理相比,与WPMY-1细胞共培养使PC3GFP细胞对奥拉帕利具有更高的耐药性(图7J)。这一效应在ADT条件下更加明显(图7J)。为了研究奥拉帕利的体内前CAF表型,下一步在NOD scid γ (NSG)小鼠中共植入PC3和WPMY-1细胞,并使用溶剂或奥拉帕利对它们进行处理(图7K)。体内奥拉帕利治疗有效降低了肿瘤生长(图7L-7M),但也产生了强烈的促结缔组织增生反应,其特征是胶原和HA沉积增强以及αSMA增加(图7N)。在内源性TRAMP小鼠模型中发现了类似的奥拉帕利诱导的促结缔组织增生性应答(图7O和7P)。为了检验这些发现与人类的相关性,将分析扩展到两个人类PCa异种移植物(VCAP和H660)和之前报告的对奥拉帕利治疗有应答的PDX模型。与小鼠数据一致,奥拉帕利在所有人类PCa模型中均诱导了强基质激活(图S6F)。
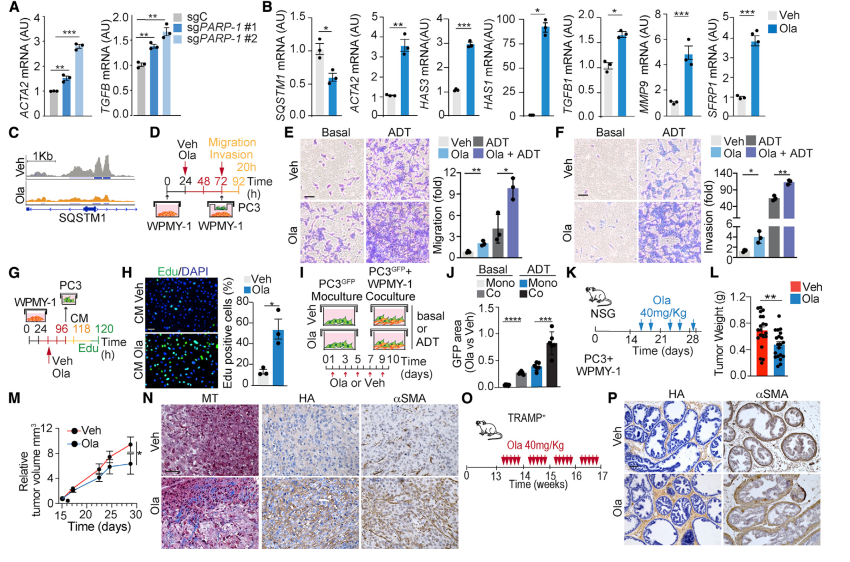
图7 奥拉帕尼治疗模拟p62的乳酸损失,促进基质激活
总结
肿瘤细胞分泌的乳酸在转录水平下调p62,从而诱导基质激活。乳酸导致NAD+/NADH比值降低,从而损害poly(ADP-ribose)polymerase 1 (PARP-1)活性。临床上使用的PARP-1抑制剂(如奥拉帕利)在下调p62和促进CAF活性方面模拟了乳酸的作用,这一事实揭示了基于PARP-1抑制剂的疗法的一个出乎意料的潜在弱点,并提示奥拉帕利与抗基质靶向疗法的联合治疗将提高奥拉帕利的疗效。