






凝血酶诱导脑缺血/再灌注过程中ACSL4依赖性铁下垂
缺血性中风对人类,特别是老年人是一个重大的危险。干预措施只能用于清除血栓,缺血性中风期间神经元死亡的机制仍存在争议。铁下垂越来越被认为是各种器官缺血后细胞死亡的一种机制。在这里,我们报道了丝氨酸蛋白酶,凝血酶,通过促进花生四烯酸的动员和随后的铁性基因,酰基辅酶a合成酶长链家族成员4 (ACSL4)的酯化来激发铁性信号传递。一种无偏多组学方法确定了凝血酶和ACSL4基因/蛋白质,以及它们的前铁毒磷脂酰乙醇胺脂质产物,在啮齿动物大脑中动脉闭塞时显著改变。在基因或药理学上抑制该途径中的多个点,可减弱体外和体内缺血模型的结果。因此,凝血酶-ACSL4轴可能是改善缺血性脑卒中铁性神经元损伤的关键治疗靶点。本文于2022年2月发表于Signal Transduction and Targeted Therapy (IF=38.104)上。
技术路线:
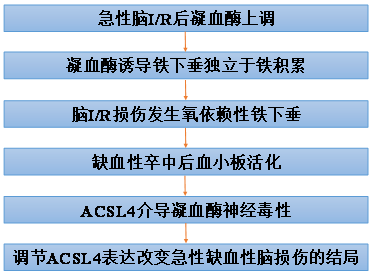
主要研究结果:
(1) 急性脑I/R后凝血酶上调
由铁下垂引起的细胞死亡是由于有毒的脂质氢过氧化物积累超过细胞解毒的能力。花生四烯酸(AA)含有磷脂酰乙醇胺(PEs),特别容易发生铁质过氧化反应。通过对小鼠缺血性中风后海马组织进行高质量的非靶向代谢组学分析(图1a),我们发现了四种磷脂显著降低,即PC(36:4)、PE(42:8)、PE (38: 6p)和PE(40:7)(图1b-h)。进一步分析其脂肪酸侧链,发现PC(36: 4,16: 0/ 20:4)、PE(42: 8,22:4 / 20:4)、PE (38: 6p, 18: 2p/ 20:4)脂肪酸侧链中均含有AA(图1i),且AA(20:4)占主导地位(图1j),这与小鼠缺血性脑卒中后海马组织游离AA显著增加(脑卒中/对照比= 1.49,p = 0.0007, Student’s t检验,图1k)一致。
为了探索缺血性卒中后AA升高的蛋白,我们对同一组织进行了蛋白质组学分析(图2a-c)。基于差异表达蛋白的富集,我们发现血小板激活、信号转导和聚集是上调最多的通路(图2d,e)。蛋白质-蛋白质相互作用(PPI)网络分析也一致指出了相同的途径(图2f),凝血酶原(F2基因编码凝血酶原,也称为凝血因子II),凝血酶原酶是上调最多的蛋白质(stroke/control ratio = 10.88, p < 0.0001, Student 's t检验,图2g),Western Blotting进一步证实了这一点(图2h)。

图1:在缺血/再灌注后,磷脂侧链上的花生四烯酸被释放
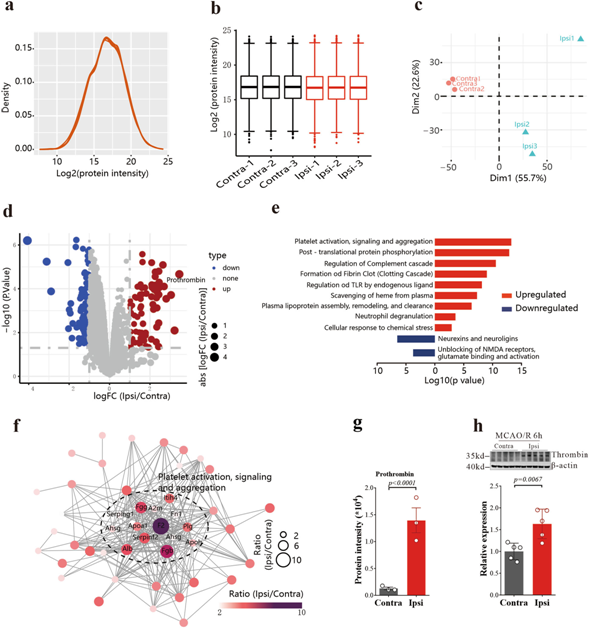
图2:急性脑缺血/再灌注后凝血酶上调
(2) 凝血酶诱导铁下垂独立于铁积累
凝血酶是缺血性脑卒中的主要药物靶点之一,其作用是促进纤维蛋白的产生和凝血。我们发现凝血酶剂量依赖性诱导N27神经元细胞死亡(图3a),并伴有AA升高(图3b)和脂质氢过氧化物升高(3c,d),线粒体变小,膜密度增加(图3e),这些特征都与铁下垂一致。凝血酶不影响细胞内亚铁的水平,这不是诱导铁下垂的必要条件(图3f)。凝血酶诱导的细胞死亡可以通过铁下垂抑制剂liproxstatin-1(图3g)和谷胱甘肽前体n-乙酰-l-半胱氨酸(图3h)和cPLA2α抑制剂darapladib(图3i)的治疗来阻止。利prox他汀-1同样可以防止MDA-MB-231细胞(一种人类乳腺癌细胞系)的细胞死亡,这表明凝血酶在脑外铁下垂中的作用(图3j,k)。
凝血酶升高是缺血性中风后AA动员的上游信号,这有助于铁下垂。达比加群在N27神经元培养的I/R氧-葡萄糖剥夺(OGD)模型中保护细胞,神经症状缓解和减少脑梗死体积(补充图未展示)。由于OGD和MCAO模型都不涉及可能与凝血酶抑制有关的血栓形成,凝血酶诱导的另一种损伤被达比加群阻断。这些数据强调了凝血酶诱导铁下垂的可能性,这可能与缺血性中风有关。

图3:凝血酶诱导神经元铁下垂
(3) 脑I/R损伤发生氧依赖性铁下垂
为了证实缺血性脑卒中中铁坠症的发生,并剖析引发铁坠症的必要条件,我们调节了MCAO/R的条件,并研究了其生化和行为变化。小鼠和大鼠在MCAO/R后(再灌注后24小时),与对侧“对照”半球相比,同侧“卒中”半球受影响的海马和皮层区域的脂质过氧化物(LPO)和丙二醛(MDA)(脂质过氧化物的分解产物)水平显著升高(小鼠:图4a,b;大鼠:补充图未展示)。透射电镜(TEM)图像显示,MCAO/R后24 h同侧皮层区域的这些特征伴有神经元线粒体收缩,铁脱铁特征(补充图未展示)。
在MCAO/R后立即给药的亚毒性剂量(没有明显的神经元损失)RSL3 (GPx4抑制剂)和erastin (Xc抑制剂)增加铁毒倾向,显著加重小鼠脑I/R损伤,表现为MDA水平显著升高(图4c),神经行为缺陷(图4d),梗死体积增加(MCAO/R后24小时;图4e)。在MCAO小鼠模型中,脂基捕获剂和原型铁脱铁抑制剂利普司他汀-1和铁司他汀-1已被证明能有效地减弱脑I/R损伤。然而,这些抑制剂不能挽救永久性MCAO(即无再灌注)造成的神经元损伤(图4f,g),因为铁上坠症依赖于氧气,进一步表明铁上坠症可能发生在再灌注阶段。
我们在I/R的氧葡萄糖剥夺(OGD)细胞培养模型中证实了神经元铁下垂的发生。N27神经元细胞经OGD处理2小时(图4h)死亡前,细胞内Fe2+水平升高(图4i),脂质过氧化物(通过BODIPY 581/591 C11 [BODIPY]测定;图4j,k),脂质氢过氧化物(Liperfluo探针),以及细胞内总ROS和MDA(补充图未展示)。与对照细胞相比,OGD处理的N27细胞中的线粒体也显著变小,但膜密度增加(补充图未展示)。利prox他汀-1治疗挽救了OGD诱导的神经元死亡(图1l),显示了铁下垂的参与。
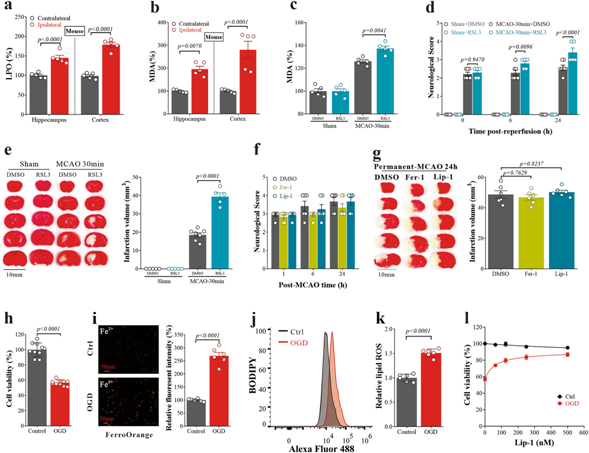
图4:脑再灌注损伤的氧依赖性铁下垂
(4) 缺血性卒中后血小板活化
凝血功能的激活是缺血性脑卒中中神经元损伤的主要原因。神经元中的凝血酶也可以通过其丝氨酸蛋白酶活性引发铁下垂。为了研究缺血性脑卒中期间凝血系统的变化,我们收集了59份血清样本,包括27例健康对照组和32例缺血性脑卒中患者,在去除血清中高丰度蛋白后,进行TMT标记的蛋白质组学分析(图5a-e)。84.7%的样本相关值大于0.98,相关值范围在0.968~0.993之间(图5a),表明结果具有可重复性。在分析中,我们总共鉴定出584个蛋白质,其中424个蛋白质可以得到≥2个多肽的支持,平均数量为8.31个(图5c)。评估了不同样本中蛋白质的分布,我们发现超过50%的样本中有362种蛋白质被定量(图5d)。基于包含362个蛋白质的随机森林估算数据的tSNE分析验证了不存在批效应(图5e)。
我们在缺血性卒中血清中共鉴定出28个蛋白上调,21个蛋白下调(图5f)。其中,缺血性脑卒中患者血清中纤维蛋白凝块形成明显上调(图5g-k),这与小鼠大脑中血小板聚集增加一致(图2f)。然而,与健康对照组相比,血清中凝血酶水平没有明显升高(图5l),这表明我们在大脑中观察到的升高(图2h)可能不是来自血液。如果达比加群具有缺血性卒中后的神经保护作用(补充图未展示),其有益作用不太可能来自于其在血液中的抑制凝血活性。相反,缺血性中风患者血液中凝血酶没有升高,这表明抗凝血酶药物的任何益处都可能是由于大脑中凝血酶的升高(图2h)。考虑到凝血酶在动员铁坠体脂肪酸AA方面的额外作用,我们假设凝血酶可能会诱导缺血性中风后的铁坠症。
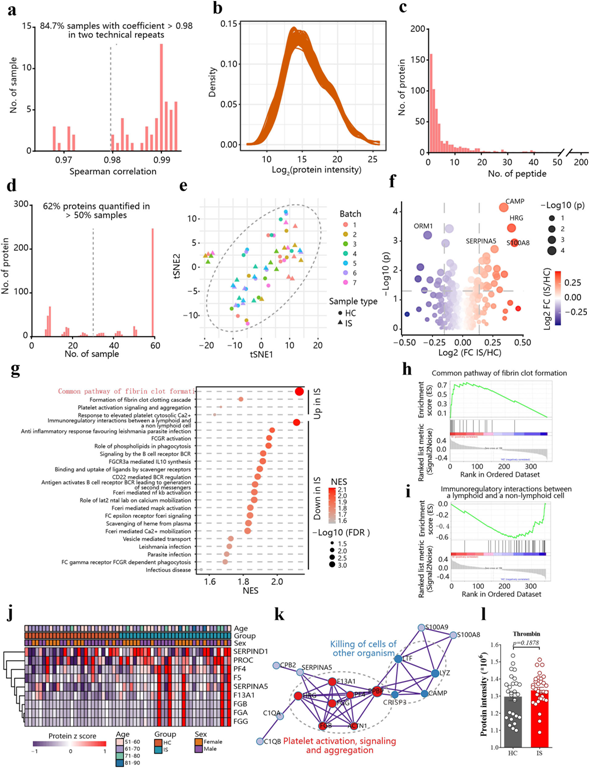
图5:缺血性卒中后血小板活化
(5) ACSL4介导凝血酶神经毒性
酰基辅酶a合成酶长链家族成员4 (ACSL4)是一种重要的脂质代谢酶,通过将游离AA转化为花生四烯醛-CoA11生成脂质氢过氧化物来参与铁脱铁。与游离AA通过促进其泛素化和蛋白酶体降解抑制ACSL4水平一致,我们发现凝血酶处理后N27细胞中的ACSL4降低(图6a),而不影响ACSL4 mRNA的表达。I/R后6 h,缺血区海马区ACSL4蛋白表达明显下降,而ACSL4 mRNA表达在同一时间点无明显变化(补充图未展示)。由于缺血性卒中后小鼠海马区游离AA显著增加(图1k),在I/R过程中ACSL4的降低可能是翻译后修饰的结果。通过Western Blot(图6b)或组织学(图6c)检测,MCAO/R后大鼠同侧海马ACSL4的表达随着时间的推移相似地下降,在I/R后3小时,ACLS4明显减少,这是在I/R后6小时开始海马神经元损失(使用NeuN评估存活神经元,使用Fluoro-Jade评估退化神经元)之前(图6d,e)。大鼠的结果与MCAO后小鼠的结果一致。然而,凝血酶在缺血早期明显升高(图6f)。这些数据表明,ACSL4下调是脑I/R的早期事件,独立于神经元死亡而发生,这可能是对凝血酶诱导应激的保护性反应。
为了阐明凝血酶细胞毒性是否由ACSL4介导,我们使用基于CRISPR-Cas9的基因编辑生成了ACSL4敲除(KO) N27细胞,使用含有ACSL4表达框的慢病毒载体mach-OE-rACSL4生成了ACSL4过表达(OE)细胞。Western Blotting证实了这两种调制(图6g,上图)。凝血酶依赖于ACSL4引起毒性,因为凝血酶的细胞毒性因ACSL4的还原而恢复,并因ACSL4的过表达而加重(图6g)。ACSL4抑制剂吡格列酮(PIO)也能阻断凝血酶的细胞毒性(补充图未展示)。这些结果表明,凝血酶可能通过促进ACSL4依赖的铁坠瘤而导致神经元细胞死亡,而ACSL4的降低可能有助于抑制凝血酶引起的铁坠瘤损伤。
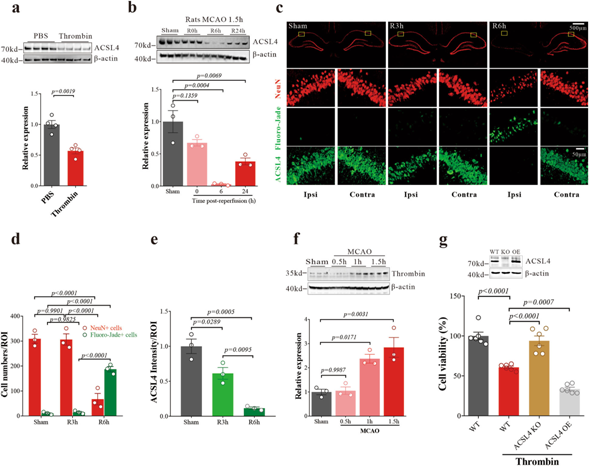
图6:凝血酶诱导急性脑缺血/再灌注后ACSL4下调
为了确定ACSL4是否影响大脑I/R后的结果,我们使用单次注射腺相关病毒载体,即AAV8-mACSL4和AAV8-EF-Cas9+AAV8-mACSL4-sp,在小鼠左海马CA3区域(小鼠MCAO/R最脆弱的区域)选择性过表达或敲除ACSL4。并检查了MCAO啮齿动物模型的影响(图7a)。以AAV8-EGFP(绿色荧光蛋白增强型AAV8)为对照。免疫荧光染色证实了转导的有效性(图7b)。在相同的30分钟MCAO/R条件下,ACSL4 OE小鼠表现出明显的梗死体积增加(灌注后24小时,图7c),神经评分恶化(图7d),旋转运动性能变差(图7e)。与EGFP小鼠相比,ACSL4 KO小鼠对I/R损伤有保护作用,梗死体积减小(图7f),神经评分(图7g)和运动协调性(图7h)也有显著改善。与铁脱落抑制剂一致,ACSL4 KO小鼠永久性MCAO后神经元损伤与对照小鼠无差异(图7i,j),这强烈提示在该模型中,铁脱落发生在再灌注期间。
激光散斑信号显示敲除ACSL4后不影响MCAO/R后大鼠皮层的血流(图7k,l),证实ACSL4 KO不是单纯通过影响血流动力学而起到保护作用。AAV辅助ACSL4敲除大鼠皮层对I/R诱导的功能损伤具有显著保护作用(神经评分;图7m)和脑梗死体积(TTC染色;图7n)。
ACSL4 KO细胞对RSL3诱导的铁坠病有保护作用,而ACSL4 OE细胞对同样的毒素脆弱(补充图未展示)。将这些细胞进行OGD/再氧化,发现ACSL4 KO细胞对OGD相关毒性具有抗性,而ACSL4 OE细胞更容易受到OGD相关毒性的影响(补充图未展示)。

结论:总之,我们的研究结果有力地证明,抗凝血酶疗法可能通过抑制铁下垂而对脑卒中再灌注后有益,也可能对涉及铁下垂的其他疾病有用。
参考文献:
Tuo, Q. Z., Liu, Y., Xiang, Z., Yan, H. F., Zou, T., Shu, Y., Ding, X. L., Zou, J. J., Xu, S., Tang, F., Gong, Y. Q., Li, X. L., Guo, Y. J., Zheng, Z. Y., Deng, A. P., Yang, Z. Z., Li, W. J., Zhang, S. T., Ayton, S., Bush, A. I., … Lei, P. (2022). Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion. Signal transduction and targeted therapy, 7(1), 59. https://doi.org/10.1038/s41392-022-00917-z.