






Science高分——METRNL调控心肌修复
心肌梗死后的有效组织修复需要在不完全定义的免疫细胞-内皮细胞相互作用的指导下,产生强有力的血管生成反应,但机制不明。本研究确定单核细胞(Mos)和巨噬细胞(Mphs)衍生的细胞因子METRNL(meteorin-like)是梗死后血管生成的驱动力和干细胞因子受体KIT(KIT受体酪氨酸激酶)的高亲和力配体。METRNL通过KIT依赖的信号传导在体外培养的人类内皮细胞中介导血管生成作用。在心肌梗死的小鼠模型中,METRNL通过选择性地扩大梗死边缘区表达KIT的内皮细胞群来促进梗死修复。Metrnl缺失的小鼠不能产生这种KIT依赖性的血管生成反应,并在梗死后出现严重的心力衰竭。本研究表明,METRNL是缺血组织修复中的KIT受体配体。本文于2022年6月发表在《Science》IF:63.714期刊上。
技术路线:

主要实验结果:
1、髓细胞来源的METRNL促进心肌梗死后血管生成
作者在急性心肌梗死小鼠模型中进行了生物信息学分泌组分析,发现了此前未鉴定到的驱动梗死修复的骨髓细胞来源的生长因子,即METRNL(meteorin-like),其在左心室梗死区域的髓系细胞中强表达(附图1)。在假手术的基线条件下,METRNL在心脏中表达较弱,但在心肌梗死后强烈表达。共聚焦显微镜显示,表达METRNL的细胞共表达髓系细胞标志物CD11b,并且定位于梗死边缘区的内皮细胞附近(图1A)。RT-qPCR显示单核细胞和巨噬细胞是梗死区域,骨髓,脾脏和外周血中主要的METRNL表达的细胞(附图2A)。Metrnl广泛表达于梗死心脏单细胞RNA测序确定的Mo和Mph簇中(附图2B)。根据CCR2和CX3CR1的表达来划分Mos和Mpsh亚群,发现METRNL在CCR2high的Mos和CCR2highCX3CR1high/low的Mphs中比在CCR2lowCX3CR1high的Mphs中表达更强(附图2C)。
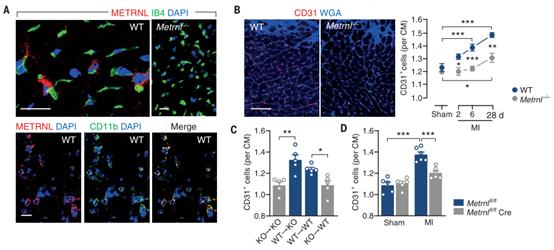
随后用Metrnl缺陷(Metrnl-/-)小鼠建立急性心肌梗死模型,探讨Metrnl在梗死修复过程中的功能。结果显示,梗死后敲除小鼠表现出更大的梗死瘢痕,伴有更明显的左室扩张和收缩功能障碍,并且梗死边缘区新生毛细血管形成受损(图1B)。在基线条件下(图1B),Metrnl-/-小鼠的心肌毛细血管密度并未降低,这表明Metrnl缺失特异性地损害了缺血损伤后的血管生成。
用WT骨髓细胞移植Metrnl-/-小鼠可逆转血管生成缺陷(图1C),并改善心肌梗死后的LV瘢痕和重塑。相反,用Metrnl-/-骨髓细胞移植WT小鼠或通过Metrnlfl/fl小鼠与LysMCre/+小鼠杂交选择性地删除骨髓细胞中的Metrn损害了梗死后的血管生成(图1C和D)并使LV瘢痕和重塑恶化。这些发现表明,骨髓细胞衍生的METRNL能促进血管生成、组织修复和心梗后的功能适应。
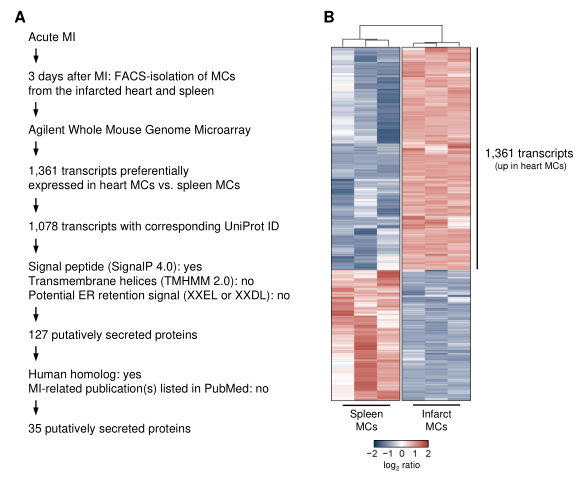
附图1生物信息学骨髓细胞分泌组筛选
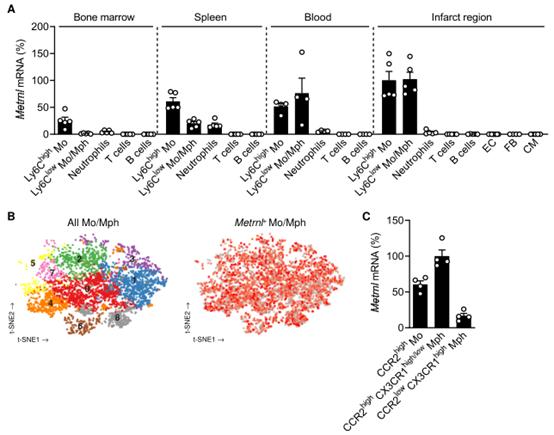
附图2 心肌梗死后不同细胞类型Metrnl mRNA的表达
2、METRNL与KIT的细胞外结构域结合
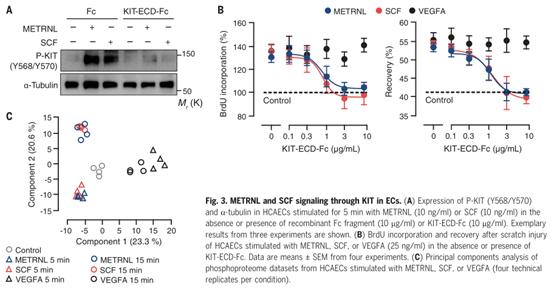
METRNL剂量依赖性地划痕损伤后的人冠状动脉内皮细胞(HCAEC)增殖和迁移(图2A-B,上),表明METRNL蛋白直接作用于ECs。使用化学交联质谱法,确定KIT是内皮细胞中METRNL细胞表面受体的候选分子。HEK-293细胞的免疫共沉淀实验证明了METRNL和KIT的结合。为评估METRNL是否与KIT的胞外结构域结合,从HEK-293细胞中制备表达METRNL、SCF、VEGFA,和/或分泌型KIT胞外结构域-Fc片段融合蛋白(KIT-ECD - Fc)的条件上清。从上清液中提取KIT-ECD-Fc,纯化了METRNL和SCF,但没有纯化VEGFA(图2A,下)。微尺度热泳动显示,METRNL和SCF以相似的高亲和力结合KIT的胞外结构域(图2B,下)。
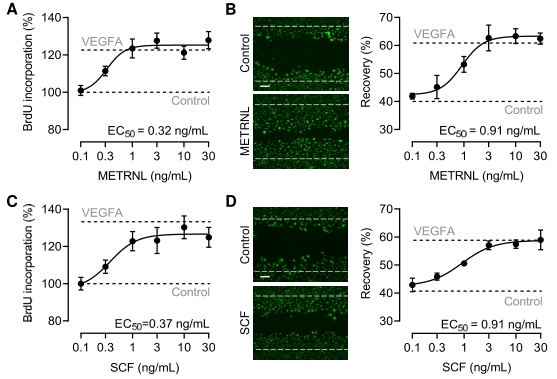
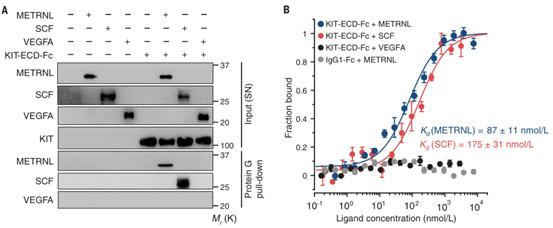
图2 METRNL与KIT的细胞外结构域结合
3、上皮细胞中METRNL和SCF信号通路通过KIT
SCF刺激可诱导受体近膜区酪氨酸残基568和570 (Y568/Y570)的KIT同二聚体化和转磷酸化,从而为SRC家族激酶创造一个对接位点。用METRNL或SCF诱导的KIT (Y568/Y570)磷酸化以相似的动力学处理HCAECs(图3A,上)。在培养基中加入重组KIT-ECD-Fc可阻止METRNL和SCF诱导KIT (Y568/Y570)磷酸化(图3A,下),这与跨膜KIT和KIT- ECD - Fc竞争与METRNL和SCF结合的观点一致。事实上,重组KIT-ECD-Fc剂量依赖性地抑制了METRNL和SCF的血管生成作用,但对VEGFA的抑制作用不明显(图3B,下)。
KIT下游,METRNL和SCF都诱导SRC(Y416)和AKT1(S473)磷酸化,同样遵循相似的信号动力学轨迹(图3B-C,上)。为了获得更广阔的视角,应用高分辨率质谱检测了METRNL、SCF或VEGFA刺激的HCAECs中的蛋白磷酸化动力学。在基于分布于1891个不同蛋白质的4750个磷酸化位点的主成分分析中,METRNL和SCF的磷酸化蛋白质组标签聚在一起,并在主成分空间中与VEGFA的磷酸化蛋白质组标签分离(图3C,下)。因此,METRNL和SCF的血管生成作用是通过KIT和共享的下游信号通路介导的。
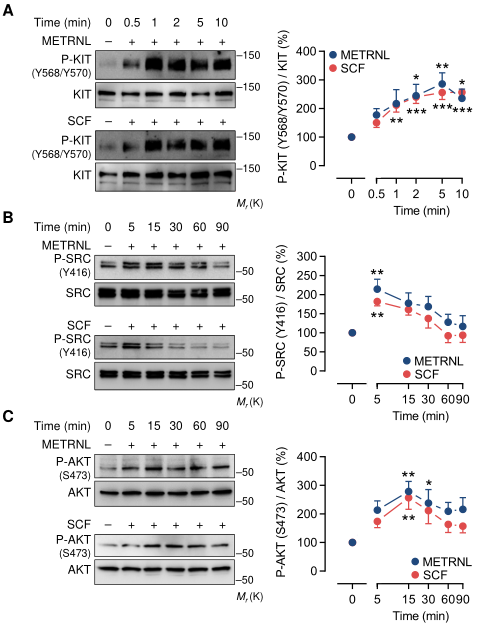
4、心肌梗死后,METRNL增加表达KIT的上皮细胞群
使用单细胞RNA测序在成年小鼠心肌中鉴定表达kit的内皮细胞。虽然在基础条件下,少数分散在几个EC簇的内皮细胞表达Kit,但在MI后,表达Kit的内皮细胞数量显著增加(图4A)。为探讨KIT表达是否会影响EC对METRNL和SCF刺激的敏感性,从c-kit H2B-tdTomato/+小鼠分离心脏内皮细胞。这些小鼠的细胞核tdTomato信号完全重现了内源性KIT的表达模式。为调查KIT在梗死边缘区的表达EC行为,用c-kit H2B-tdTomato/+小鼠构建急性心肌梗死模型。正如单细胞RNA测序观察到的,在基线条件下和心肌梗死后,c-kit H2B-tdTomato阴性的内皮细胞数量超过c-kit H2B-tdTomato阳性的内皮细胞(图4B)。然而,两个人群的扩张对梗死边缘区的毛细血管密度增加的贡献大致相同(图4B)。
与METRNL不同,SCF由梗死区域的ECs、成纤维细胞和心肌细胞表达(附图3A),并且MI后SCF的表达没有增加(附图3B)。此外,在Metrnl- / -小鼠的梗死区域未观察到SCF上调(附图3C)。为研究METRNL是否驱动KIT在心肌梗死后EC中的表达,将c-kit H2B-tdTomato等位基因交叉到METRNL- / -小鼠中。在基线条件下,Metrnl缺失不影响c-kit H2B-tdTomato阳性或阴性的心肌EC密度(图4C)。这表明在发育过程中,METRNL对于建立细胞群都不是必需的。然而,心肌梗死后,Metrnl- / -小鼠的c-kit H2B-tdTomato阳性的EC数量没有增加,而c-kit H2B-tdTomato阴性EC扩增不受影响(图4C)。
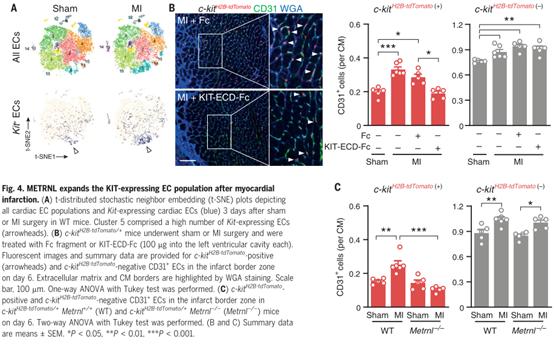
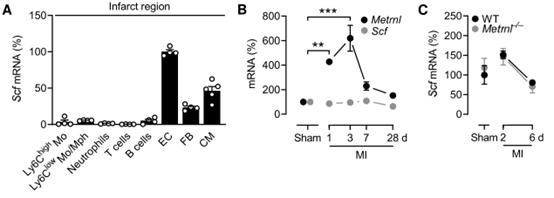
附图3心肌梗死后SCF在心脏的表达
参考文献:
RebollMR, Klede S, Taft MH, Cai CL, Field LJ, Lavine KJ, Koenig AL, Fleischauer J, Meyer J, Schambach A, NiessenHW, Kosanke M, van den Heuvel J, Pich A, Bauersachs J, Wu X, Zheng L, Wang Y, Korf-Klingebiel M, Polten F, Wollert KC. Meteorin-like promotes heart repair through endothelial KIT receptor tyrosine kinase. Science. 2022 Jun 17;376(6599):1343-1347. doi: 10.1126/science.abn3027. Epub 2022 Jun 16. PMID: 35709278; PMCID: PMC9838878.