






MicroRNA-210在心肌梗死中控制线粒体代谢并保护心脏功能
尽管最近在急性心肌梗塞(AMI)的治疗和患者生存率的改善方面取得了进展,但缺血性心脏病仍然是全球死亡的主要原因,也是慢性心力衰竭的主要原因。仍然需要新的心脏保护策略来减轻AMI的有害影响,并改善冠心病患者的不良心脏重塑和心功能障碍。动物研究表明,microRNA 210(miR-210)是治疗缺血性心脏病的潜在疗法,以改善心肌梗死小鼠模型中的心脏功能。人体临床研究表明miR-210是冠状动脉疾病的生物标志物。MiR-210是细胞缺氧反应的主要调节因子,通过靶向线粒体能量代谢和活性氧(ROS)通量。线粒体除了通过氧化磷酸化是收缩细胞ATP的主要来源外,还是细胞ROS产生的关键来源。在缺血再灌注(IR)的情况下重新编程线粒体代谢和ROS通量是心脏损伤的关键驱动因素。因此,在IR期间抑制线粒体呼吸链和减少线粒体耗氧量可保护缺血性心脏病。该研究发表于《Circulation》,IF: 39.918。
技术路线:

主要研究结果:
1. MiR-210缺乏以性别依赖的方式加重IR诱导的雄性小鼠心功能障碍
MiR-210敲除小鼠和其野生型对照C57Bl/6小鼠在2月龄时通过结扎冠状动脉左中前降支30分钟后再灌注的方法建立在体心脏缺血再灌注模型。未结扎左冠状动脉前降支的假手术动物作为对照组。分别于再灌注4 h和24 h后分离心脏,检测心脏中miR-210的表达。如图1A所示,在WT小鼠的心脏中,miR-210在两个时间点都显著增加,而KO小鼠没有。在女性和男性基线之间以及在女性和男性IR治疗之间没有发现miR-210水平的显著差异。在体内IR治疗前和治疗后6周,通过超声心动图评估心功能。如图1B至1E和图S1A至S1G所示,在基线和假手术动物中,miR-210 KO和WT小鼠的超声心动图无显著差异。IR导致雄性WT小鼠室间隔舒张末期、收缩末期、舒张末期左心室后壁厚度、收缩末期左心室后壁厚度、射血分数(EF)、短轴缩短率(FS)降低,左心室舒张末期内径、收缩末期内径、舒张末期容积、收缩末期容积增加。在雌性WT小鼠中,IR降低EF和FS,增加左室收缩末期内径和收缩末期容积。值得关注的是,miR-210的缺失揭示了显著的性别差异,并显著加重了IR诱导的雄性小鼠心脏功能障碍,而不影响雌性动物。
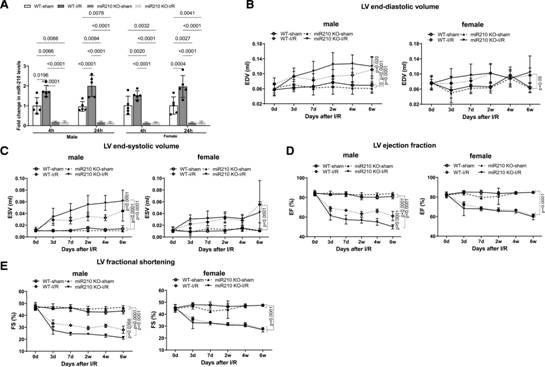
图1MiR-210缺陷加重雄性小鼠心肌梗死后的心功能障碍
2. MiR-210模拟物挽救了MiR-210缺乏对ir诱导的心肌梗死和心功能障碍的作用
鉴于miR-210缺陷仅影响雄性小鼠,作者接下来的研究将集中在雄性动物。作者发现,与野生型对照相比,在急性IR损伤的情况下,miR-210缺乏增加了MI体积,而miR-210模拟物挽救了miR-210缺乏对MI的影响(图2A和2B)。同样,miR-210缺陷增强了LV EF和FS的功能障碍,而miR-210模拟物阻断了miR-210 KO小鼠中过度的功能障碍(图2C和图2D)。
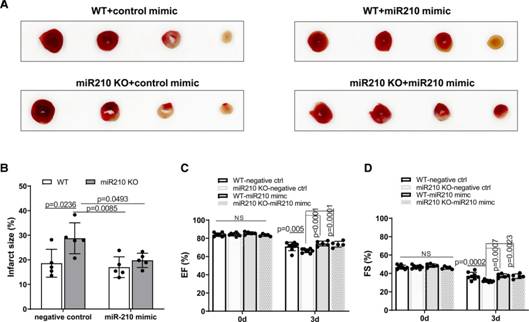
图2MiR-210 mimic对缺血再灌注诱导的心肌梗死和心功能障碍具有保护作用
3. MiR-210在急性心脏IR中控制线粒体生物能学
线粒体是miR-210在细胞缺氧反应中的主要靶点。在缺血再灌注过程中抑制线粒体耗氧量和生物能对缺血性心脏病具有保护作用。因此,作者研究了miR-210在急性心脏IR中控制线粒体生物能学的作用。与此同时,作者进行了Seahorse通量分析,以评估离体心肌纤维束的氧消耗率(OCR,氧化磷酸化指数)和乳酸生成(通过细胞外酸化率(ECAR,糖酵解指数)评估。Seahorse测量的OCR和ECAR的实时轨迹和平均数据如图3A和3G所示。在心肌缺血30分钟和再灌注24小时后,作者观察到,与WT动物相比,miR-210 KO小鼠心脏的基础OCR和最大呼吸能力显著且可重复地增加(图3B和图3C)。然而,与与质子泄漏相关的OCR显著增加(图3E)相比,用于ATP生成的氧使用量显著减少(图3D),从而导致能量生成的耦合效率显著降低(图3F)。与此同时,作者观察到miR-210 KO小鼠的基础ECAR和糖酵解能力降低(图3H和3I)。miR-210缺陷对心脏线粒体生物能学的这些影响可通过miR-210模拟物恢复(图3A-3I)。这些发现明确表明miR-210促进线粒体代谢从氧化磷酸化转变为糖酵解,从而微调心脏对急性IR和MI的反应。
4. MiR-210在急性心脏IR损伤和心功能障碍中抑制线粒体ROS
在心脏缺血再灌注的情况下,miR-210可能作为一种稳态机制来抑制线粒体生物能学和氧消耗,缓解电子传递和氧浓度的不匹配,并降低线粒体ROS (mtROS)通量。接下来,作者使用miR-210模拟物和miR-210 KO小鼠,通过功能获得和功能丧失的方法,研究了miR-210在IR期间调节心脏mtROS生成中的机制联系。MiR-210 KO和WT对照组小鼠采用缺血30 min再灌注24 h的方法,采用mtROS特异性荧光探针MitoSOX.20检测心脏线粒体来源的ROS作者发现,在IR处理前1小时静脉注射miR-210模拟物可阻断KO小鼠心脏中mtROS的增加(图4A)。与此一致,作者观察到,在IR处理前15分钟静脉给予线粒体特异性抗氧化剂MitoQ (4 mg/kg)降低了WT小鼠心脏中的mtROS,并否定了miR-210 KO小鼠中增加的mtROS(图4B)。然后,作者研究了MitoQ是否以及在多大程度上可以恢复miR-210缺陷对ir诱导的心肌梗死和心功能障碍的影响。作者发现,MitoQ减少了WT小鼠中IR诱导的MI,并否定了miR-210缺陷对IR介导的MI的影响(图4C和4D)。与此同时,作者观察到,在IR后3天的miR-210 KO小鼠中,MitoQ挽救了miR-210缺乏对LV EF和FS过度功能障碍的影响(图4E和4F)。
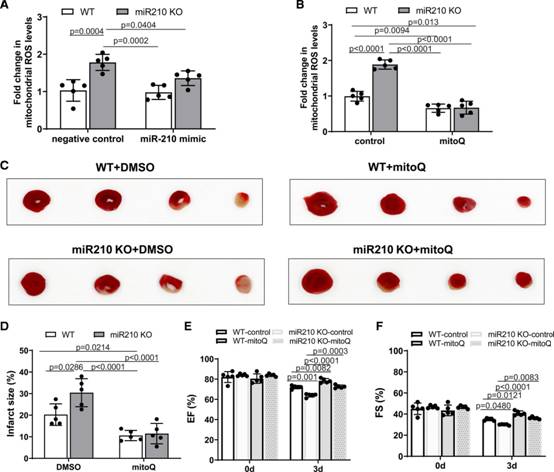
图4MiR-210抑制线粒体ROS在急性心脏缺血再灌注损伤和心功能障碍中发挥作用
接下来,作者检测了通过MitoQ抑制mtROS的产生对WT和miR-210 KO小鼠心脏线粒体呼吸和糖酵解的影响。作者观察到,在急性IR环境下,MitoQ对WT小鼠的基础线粒体OCR和最大呼吸能力没有显著影响,但阻断了miR-210缺陷对线粒体OCR的影响(图5B和图5C)。在WT动物的心脏中,MitoQ显著增加了用于ATP生成的氧使用量,降低了与质子泄漏相关的OCR,并消除了miR-210缺乏的影响(图5D和5E)。与此同时,尽管MitoQ抑制了线粒体呼吸,但它显著提高了能量产生的耦合效率(图5F)。此外,作者发现MitoQ降低了WT小鼠的基础ECAR和糖酵解能力,并否定了miR-210缺乏对急性IR环境下心脏糖酵解的影响(图5H和5I)。综上所述,这些发现表明MitoQ的保护作用是通过其对急性IR时心脏线粒体呼吸和mtROS生成的影响以及对糖酵解应激的释放来介导的。

图5MitoQ挽救了miR-210缺乏对线粒体生物能学的影响
5. GPD2是miR-210控制心肌细胞mtROS的一个新靶点
作者首先研究了miR-210靶向GPD2转录本3 ' -非翻译区(3 ' -UTR)的作用。图6A描绘了miR-210靶向位点的GPD2 3 ' -UTR序列,以及miR-210沉默GPD2 mRNA翻译的示意图。作者使用序列对比程序MultAlin (http://multalin.toulouse.inra.fr/multalin/multalin.html)在GPD2 3 ' -UTR中确定了一个潜在的miR-210互补结合位点,并使用rna诱导沉默复合物免疫沉淀实验证明,与阴性对照处理的细胞相比,在miR-210模拟物转染的小鼠新生心肌细胞中,GPD2 3 ' -UTR富集约5倍(图6B),这表明GPD2是miR-210的直接靶点。然后,作者在体外氧糖剥夺(OGD)模型中评估GPD2在miR -210介导的心肌细胞保护中的作用。作者首先证实,1% O2 OGD 2小时和复氧24小时显著上调心肌细胞中miR-210的水平,这一上调被miR-210 LNA(图6C)。作者发现,在ogd处理的心肌细胞中,miR-210 LNA抑制miR-210可上调GPD2蛋白丰度,而GPD2小干扰RNA处理可逆转这一作用(图6D)。miR-210 LNA显著增强了OGD/复氧诱导的心肌细胞损伤和乳酸脱氢酶释放,用小干扰rna敲低GPD2可抵消这一作用(图6E),揭示了GPD2下调在miR-210介导的心肌细胞缺氧损伤保护中的作用。接下来,作者观察到,与健康对照组相比,miR-210 LNA的存在增加了OGD/复氧处理的心肌细胞的mtROS(图6F)。作者发现,敲低GPD2减少了OGD/复氧诱导的mtROS,并否定了miR-210 LNA对OGD/复氧介导的mtROS增加的作用(图6F)。由于GPD2敲低抵消了miR-210抑制对mtROS生成增加和心肌细胞OGD/复氧损伤的影响,作者随后研究了GPD2敲低对心肌细胞线粒体呼吸和糖酵解的影响。作者观察到,在暴露于OGD/复氧的心肌细胞中,GPD2敲低对非线粒体呼吸和备用呼吸能力没有显著影响(数据未显示),但阻断了miR-210抑制对线粒体OCR的影响(图6G-6I)。此外,在OGD/复氧处理的心肌细胞中,GPD2敲低显著增加了ATP生成过程中的氧使用,降低了与质子泄漏相关的OCR,提高了线粒体能量生成的偶联效率,并抵消了miR-210 LNA诱导的效应(图6J-6L)。同样,作者发现GPD2敲低降低了OGD/复氧心肌细胞的糖酵解和糖酵解能力,并抵消了miR-210抑制的作用(图6M-6O)。
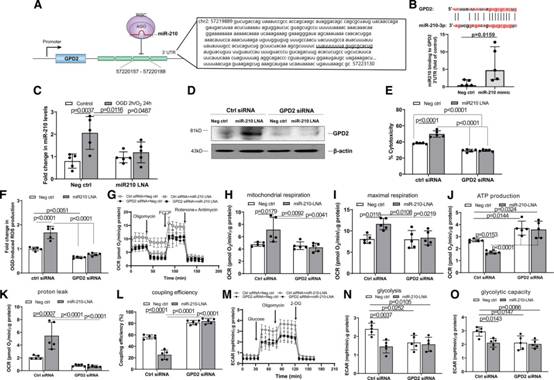
图6 MiR-210靶向GPD2抑制线粒体生物能学调节心肌细胞缺氧损伤
接下来,作者研究了MitoQ对心肌细胞缺氧损伤中miR210-GPD2-mtROS信号通路的线粒体的具体作用。作者发现,在ogd处理的心肌细胞中,用miR-210 LNA抑制miR-210显著增加了线粒体中的ROS,这一过程被MitoQ特异性阻断(图7A和7B)。与之相对应,MitoQ否定了miR-210 lna介导的OGD/复氧诱导的心肌细胞损伤增加(图7C),揭示了心肌细胞中miR-210信号通路中MitoQ作用于mtROS的机制联系。鉴于miR-210 LNA在ogd处理的心肌细胞中上调GPD2蛋白丰度,作者随后评估GPD2过表达对心肌细胞mtROS的具体影响。作者观察到,与miR-210 LNA类似,GPD2过表达增加了OGD/复氧诱导的线粒体和心肌细胞损伤中的ROS,这一过程被MitoQ阻断(图7D-7F)。此外,作者发现在OGD/复氧处理的心肌细胞中,GPD2过表达增强了线粒体OCR,降低了ATP生成的氧耗量,增加了与质子泄漏相关的OCR,降低了线粒体能量生成的偶联效率,并降低了糖酵解和糖酵解能力。重要的是,MitoQ消除了GPD2过表达诱导的效应(图S8)。因此,功能缺失和功能获得的研究结果提供了明确的证据,表明GPD2在miR-210对心肌细胞mtROS生成的影响中起关键作用。
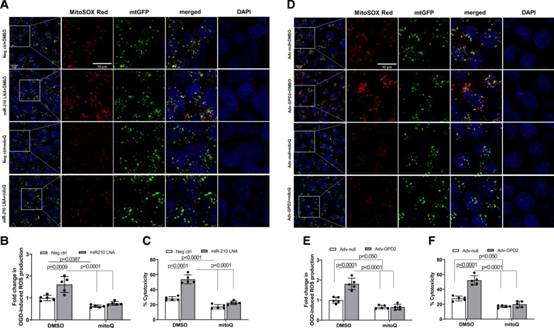
图7MitoQ抑制miR-210 LNA和GPD2过表达诱导的mtROS生成和心肌细胞缺氧损伤
6. 敲低GPD2保护心脏并否定miR-210缺乏对IR损伤的影响
然后,作者确定了内源性miR-210是否以及在多大程度上调控心脏中的GPD2。作者发现,与WT小鼠相比,miR-210 KO小鼠心脏中GPD2蛋白丰度显著增加(图8A)。miR-210 KO小鼠IR后4小时和24小时心脏中GPD2的增加分别维持和增强(图8B)。作者在MI中验证了GPD2是miR-210的靶基因,并在缺血30分钟再灌注24小时的心脏中证明了内源性miR-210与GPD2的3 ' -UTR结合。在miR-210 mimic转染的心肌细胞中,miR-210与GPD2 3 ' -UTR的结合增加,这与miR-210 mimic转染的心肌细胞中miR-210与GPD2 3 ' -UTR的结合增加一致,IR显著增加了心脏中miR-210与GPD2 3 ' -UTR的结合,而这在miR-210 KO小鼠中未观察到(图8C)。与此同时,在IR处理前静脉给予miR-210模拟物降低了WT小鼠心脏中的GPD2蛋白丰度,并阻断了miR-210缺乏对急性心脏IR中GPD2蛋白的影响(图8D)。
在WT和miR-210 KO小鼠中,作者进一步确定敲低GPD2是否以及在多大程度上抑制了ir诱导的mtROS生成和MI。作者首先验证了在接受急性IR治疗的WT和miR-210 KO小鼠的心脏中,GPD2小干扰rna敲低GPD2蛋白的表达(图8E)。然后,作者观察到,在WT和KO小鼠中,敲低GPD2蛋白丰度降低了ir诱导的mtROS生成和MI,并否定了miR-210缺陷对增加mtROS生成和MI的影响(图8F-8H)。与此同时,在心脏中敲低GPD2恢复了IR后3天miR-210 KO小鼠中观察到的过度的LV EF和FS功能障碍(图8I和8J)。
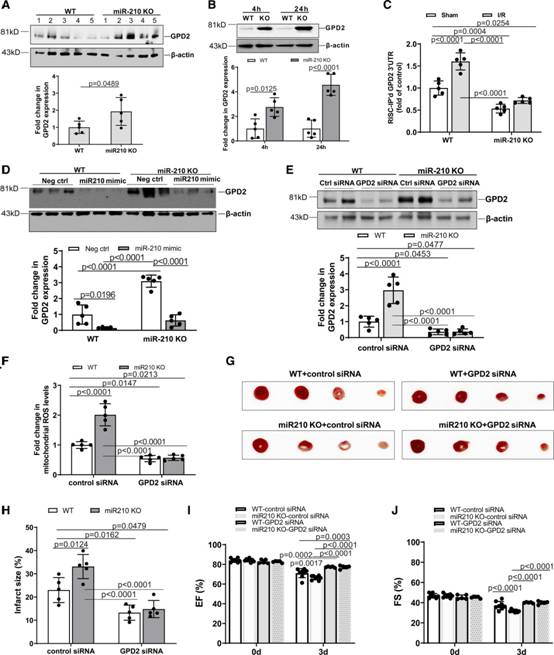
图8敲低GPD2保护心脏,并抵消miR-210缺乏对IR损伤的影响
结论:
该研究在miR-210 KO小鼠中明确证明了内源性miR-210在心肌梗死小鼠模型中保护心脏和改善心功能的作用。功能缺失和功能获得的研究结果提供了明确的证据,表明miR-210在控制线粒体生物能学和ROS生成方面是必要和充分的,并支持miR-210在心脏IR损伤中保护心肌细胞能量的有益作用。该研究确定了GPD2在miR -210介导的心脏mtROS生成调控中的一个新的机制链接。该研究还揭示了MitoQ在抑制mtROS和维持线粒体ATP生成方面的复杂作用,并展示了MitoQ在治疗缺血性心脏病和心力衰竭方面的治疗潜力。
参考文献:
Song R, Dasgupta C, Mulder C, Zhang L. MicroRNA-210 Controls Mitochondrial Metabolism and Protects Heart Function in Myocardial Infarction. Circulation. 2022 Apr 12;145(15):1140-1153.