






MYC和MET协同驱动具有不同分子特征和脆弱性的肝细胞癌
转录因子MYC和受体酪氨酸激酶MET的激活增强是肝细胞癌(HCC)中经常发生的事件之一。这两个基因各自都是肝癌发生和发展的驱动因素。然而它们在HCC中的伴随改变还未被探索。我们分析了五个独立的人类HCC队列的数据库,发现了一个以预后不良为特征的高水平MYC和MET (MYChigh/METhigh)患者的子集。这一临床观察促使我们探索MYC和MET在体内共存的功能,结合水动力尾静脉注射在R26stopMet遗传背景下的MYC表达,其中野生型MET水平在基因缺失后增强。结果表明,即使在没有与慢性疾病状态相关的预先存在的损伤的情况下,肝细胞中MYC和MET表达的增加也足以诱导肝脏肿瘤的发生。MET肿瘤中的异位MYC增加了Mki67增殖标记物的表达,并将其转化为Afp、Spp1、Gpc3、Epcam的缺失,同时Hgma1、Vim和Hep-Par1水平的升高。我们还发现了特异性免疫检查点表达的开关,Ctla-4和Lag3淋巴细胞共抑制反应增加,肿瘤细胞的Icosl共刺激反应增加。我们提供了一些人HCC细胞系对联合MYC和MET靶向的脆弱性的体外证据,这些细胞系对单一抑制具有抗性。机制上,MYC和MET的联合阻塞将由MYC或MET单独阻塞引发的部分细胞抑制作用转化为细胞毒性作用。这些发现突出了以MYChigh/METhigh为特征的HCC亚群,并记录了MYC和MET在肝脏肿瘤发生中的功能协同作用。MYC-R26Met模型是HCC生物学、患者分类和治疗的相关设置。本文于2022年11月发表于Cell Death and Disease(IF=9.685)。
技术路线:
结果:
在发育过程中MYC的过表达可以驱动HCC的形成,但通过水动力尾静脉注射在C57BL6中表达MYC只会与其他致癌驱动因素联合导致肿瘤。我们探讨了MYC和MET的高表达水平是否在五种不同的人类HCC队列中同时发生。LIRI-JP队列中,236例MYChigh/METhighHCC患者中有40例(16.9%)(图1A)。在另外两个独立的HCC患者队列中,81例患者中有13例和32个中的11个为MYChigh/METhigh(图1A)。在TCGA队列中发现了较小比例的MYChigh/METhighHCC患者(2.7%;图1A)。在所有HCC分析患者中,约18.8%为MYChigh/METhigh。
我们分析了MYChigh/METhighHCC患者亚群的临床特征。与MYClow/METhigh相比,患者的总生存期更短(图1B)。与MYClow/METhighHCC患者相比,MYChigh/METhighHCC患者没有分子特征、病因或突变。
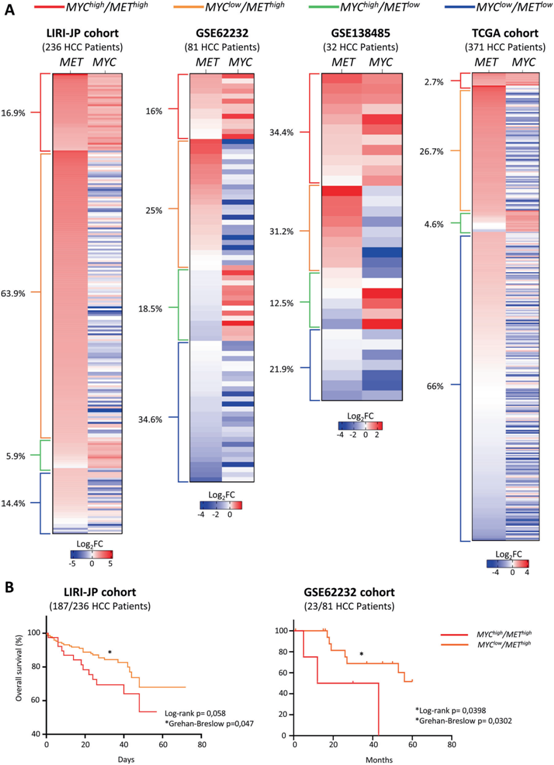
图1:MYC和MET的高表达水平在一部分HCC患者中同时发生
(2) 在小鼠肝细胞亚群中MYC和MET的同时上调引发肿瘤发生
我们评估了高MYC和MET水平在HCC患者中同时存在是否在功能上与肝癌的发生有关。我们推断R26stopMet基因设置中的强制MYC表达可能是建模该患者亚组的合适系统。我们进行了流体动力尾静脉注射两个质粒(a)短暂表达Cre重组酶,通过删除一个停止盒(Cre质粒)允许METtg表达;(b)睡美人转座酶的瞬时表达触发Myc转基因基因的基因组插入(Myc质粒;图2A)。两组对照组(单独使用Cre或Myc质粒)随访24周后,均未出现任何肉眼可见的肿瘤发生迹象(图2B, C)。相反,11/12只MYC-R26Met小鼠(通过尾静脉注射同时使用Cre和MYC质粒产生)发生肿瘤(图2B-D)。肿瘤重量在0.39-1.21 g之间,另外一个大肿瘤达到3.13 g(图2E)。逆转录-定量聚合酶链式反应(RT-qPCR)分析证实MYC-R26Met与Alb-R26Met肿瘤相比MYC表达上调(图2F)。在MYCR26Met和ALB-R26Met肿瘤中都发现了相似的METtg表达水平(图2F)。这些结果表明MYC和MET共同触发小鼠肝脏肿瘤的发生。

图2:在小鼠肝细胞亚群中MYC和MET的同时上调引发肿瘤发生
(3) 在MET癌模型中MYC上调改变HCC的分子特性
病理学家复查苏木精/伊红染色,证实肿瘤具有中分化至高分化HCC的特征,由实体或小梁状排列的多边形肿瘤细胞组成(图3A)。Ki67免疫荧光染色显示MYC-R26Met肿瘤的增殖指数明显高于Alb-R26Met肿瘤(图3B, C)。通过RT-qPCR分析Mki67mRNA水平证实,MYC-R26Met肿瘤的增殖率明显高于Alb-R26Met肿瘤(图3D)。
为了进一步表征MYC-R26Met肿瘤,我们通过RT-qPCR分析了一系列标记物的mRNA水平。Afp(未分化HCC的标记物)、Spp1、Gpc3(早期HCC的标记物)和Epcam(干性标记物;图3E, H),表明MYC-R26Met肿瘤可能比Alb-R26Met肿瘤分化更大,处于更晚期。MYC-R26Met与Alb-R26Met肿瘤中与HCC特征相关的其他标志物(Saa1, Fabp1)、祖细胞(Hnf4a, Krt19)、Wnt通路(Glul, Lgr5, Oat)、代谢(Ark1b10, Gpx2)和分化标志物(Arg1, Ctlc, Hsp1, Yap1)的mRNA表达无任何差异(图3H)。
我们研究了HMGA1的作用,HMGA1是一种非组蛋白染色质相关蛋白,最近被描述为MYC阴性三阴性乳腺癌中过表达的标记物。HMGA1是一种有效的致癌基因,可引发肿瘤进展,并与未分化的茎样表型和侵袭性有关。HMGA1是依赖MYC促进茎干性和上皮-间充质转化的正反馈循环的一部分。与Alb-R26Met肿瘤相比,Hgma1在MYC-R26Met肿瘤中上调(图3F, H),这与最近报道的Hgma1参与MYC调控一致。
为了进一步描述表型转化,我们分析了描述良好的上皮-间充质转化(EMT)标记的mRNA水平,并与MYC促进实体肿瘤中EMT的能力相关。与Alb-R26Met肿瘤相比,MYC-R26Met中Vimentin(Vim)间充质标记物增加,E-cadherin (Cdh1)上皮标记物减少(图3G, H),表明MYC-R26Met具有更多的间充质表型,这与侵袭性、预后差和对目前临床使用的药物的耐药性有关。我们进行了免疫荧光分析,以进一步记录MYC-R26Met与Alb-R26Met肿瘤中发现的分子开关。MET在Alb-R26Met和MYC-R26Met肿瘤中均有表达,而MYC高水平仅限于MYC-R26Met肿瘤(图4A, B)。MYC-R26Met与Alb-R26Met肿瘤相比,AFP和OPN减少,HMGA1和Hep-Par1染色增加(图4A, B)。
最近的研究将MYC描述为不同实体癌中免疫微环境的重塑者。我们通过RT-qPCR探索MYC-R26Met肿瘤与Alb-R26Met肿瘤的免疫检查点是否存在切换。相比于Alb-R26Met肿瘤,MYC-R26Met中Ctla4和Lag3表达更高(图5A, B)。在MYC-R26Met和Alb-R26Met肿瘤中,Icosl mRNA水平有轻微增加,但其受体Icos无增加,这表明假定存在共刺激反应(图5C)。
我们研究了MET肿瘤中MYC过表达后基因表达的转换是否发生在其他小鼠模型中,在这些模型中,HCC是由流体动力学共注射质粒驱动MYC表达与不同已知癌基因(GSE148379)引发的。MYC与MET以外的癌基因的过表达并没有导致MYC-R26Met与Alb-R26Met肿瘤中切换基因的表达发生显著变化(Afp, Spp1, Gpc3, Epcam, Mki67, Hgma1, Csp1 (Hep-Par1),Vim,Cdh1标记物,Ctla4, Lag3, Icosl免疫检查点;图5D)。这些发现揭示了当MYC过表达在MET上调的情况下发生时,特定标记物水平的有趣切换。
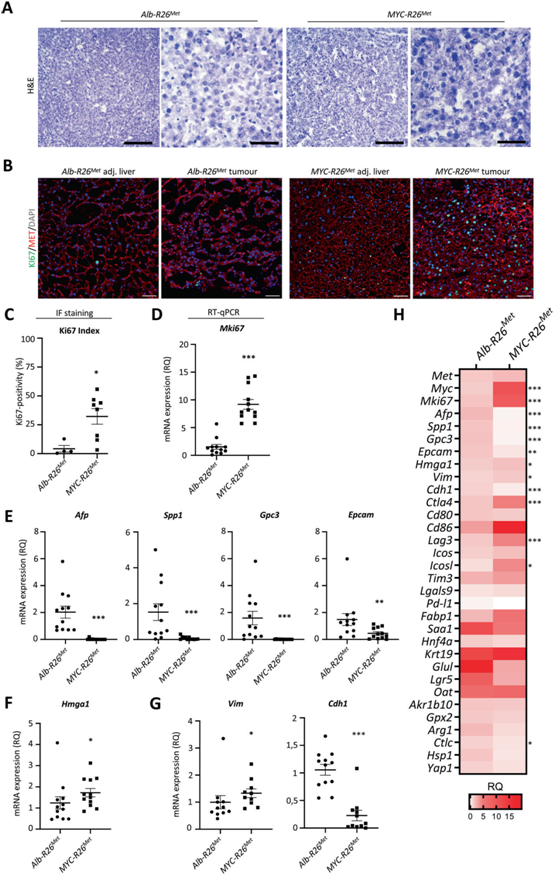
图3:Myc-R26Met和Alb-R26Met肿瘤的肝细胞特征
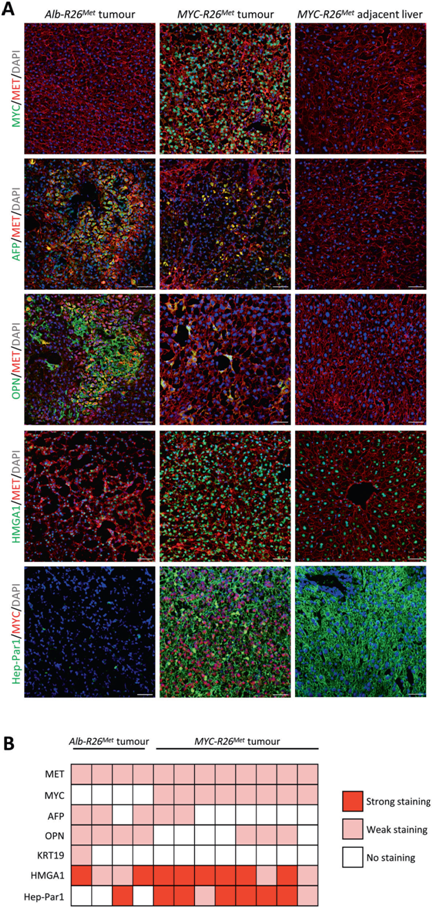
图4:Myc-R26Met和Alb-R26Met肿瘤的分子特征
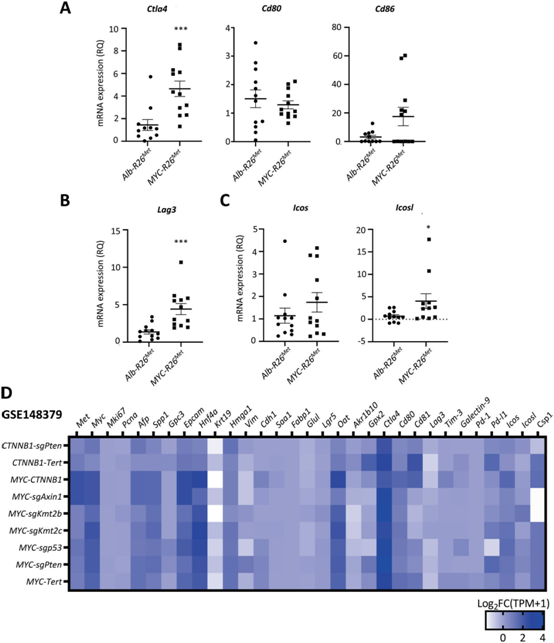
图5:MYC上调改变MET癌模型中HCC的免疫相关分子特性
(4) MYC和MET的组合靶向在人类HCC细胞亚群中给予反应性
根据转录组学数据,一组人肝癌细胞系先前已被细分为三个亚群。CL1亚群对应于大多数分化的细胞。CL3对应于分化较低的细胞,具有间充质特征,以及侵袭性和干细胞样标记物。CL2亚群对应于具有混合上皮-间充质、肝特异性和干细胞样特征的细胞。MYC和MET水平之间没有发现相关性,只有CL3亚组有不显著的趋势。我们选择了CL1和CL3人类HCC细胞的一个子集来分析细胞提取物中的MYC和MET蛋白水平。细胞在MYC和MET水平上表达程度略有不同(图6A, B),无明显相关性。JHH5细胞的特征是MET磷酸化和下游GAB1信号的激活,与HGF的表达一致以及自分泌MET激活(图6A)。
我们选择了三种人HCC细胞系(JHH5、Hep3B和Huh7),涵盖了一系列MYC和MET水平,以评估细胞对其靶向反应的活力。使用10058-F4抑制MYC,其干扰MYC-MAX相互作用并阻止MYC靶基因表达的转激活。10058-F4以剂量依赖的方式降低了测试的HCC细胞的活力(图6C)。cabozantinib是临床用于HCC治疗的一种多RTK抑制剂,它对MET的抑制仅部分干扰了我们测试的人类HCC细胞的活力(图6C)。阻断MYC和MET这两种药物的联合使用显著降低了我们测试的人类HCC细胞的活力(图6C)。我们使用了另一种MYC靶向剂Omomyc,一种在癌细胞中作为阻断MYC功能的显性阴性剂的肽。处理72 h后,仅在Omomyc存在的人类Hep3B细胞上观察到活性降低(图6D)。与单一药物相比,Omomyc和cabozantinib联合用药显著降低了细胞存活率(图6D)。生化实验证实了10058-F4和Omomyc对MYC转录功能的影响,例如SURVIVIN、MYC和CYCLIND1的下调,尽管分析的细胞系之间存在差异,并且与使用的MYC阻滞剂有关(图6E, F)。MET水平无重大变化(图6E,F),这与HCC细胞在联合使用MYC阻滞剂时对cabozantinib保持敏感性一致。我们还评估了MYC和MET单独和联合靶向HLE和HLF细胞的效果,这些细胞被归类为CL3亚类。细胞活力测定证实了MYC和MET联合靶向与单一治疗相比的效力(图7A),生化研究证实了MYC靶向后SURVIVIN、MYC和CYCLIND1的下调(图7B)。
为了从机制上探索单一和联合处理之间的差异,我们在Huh7细胞中使用抗磷酸化组蛋白H3和抗切割Caspase3检测细胞增殖和凋亡。10058-F4或cabozantinib单药治疗可轻微降低HCC细胞的增殖率,而不触发切割Caspase3的表达(图8A, B)。相比之下,10058-F4和cabozantinib联合治疗可诱导细胞凋亡(图8A, B)。这些结果表明,MYC靶向使HCC细胞对cabozantinib易感,将部分细胞抑制作用(由单一治疗触发)转化为细胞毒性作用(由联合治疗实现)。
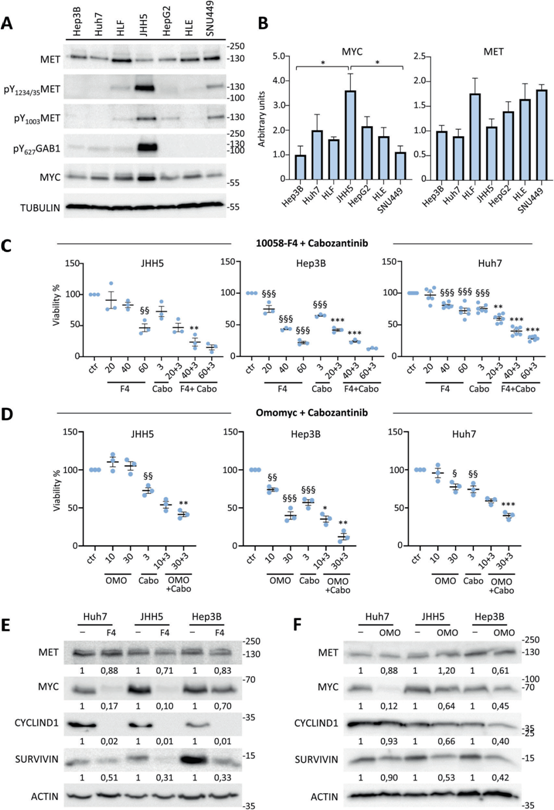
图6:MYC靶向赋予HCC细胞系亚群对cabozantinib治疗的反应性
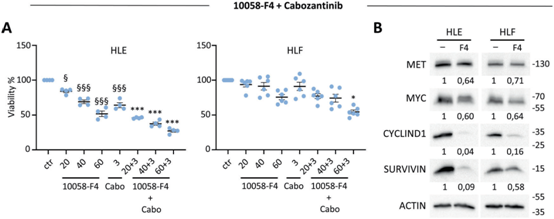
图7:MYC靶向使CL3亚组的HCC细胞系对cabozantinib治疗敏感

图8:MYC和MET的联合阻断将轻度细胞抑制作用转变为强烈细胞毒性作用
结论:
MYC阻塞通过降低MYC靶点的表达水平使HCC细胞对cabozantinib的反应性增加,从而提供了对MET信号支持的更高程度的依赖。
参考文献:
Sequera, C., Grattarola, M., Holczbauer, A., Dono, R., Pizzimenti, S., Barrera, G., Wangensteen, K. J., &Maina, F. (2022). MYC and MET cooperatively drive hepatocellular carcinoma with distinct molecular traits and vulnerabilities. Cell death & disease, 13(11), 994. https://doi.org/10.1038/s41419-022-05411-6.