






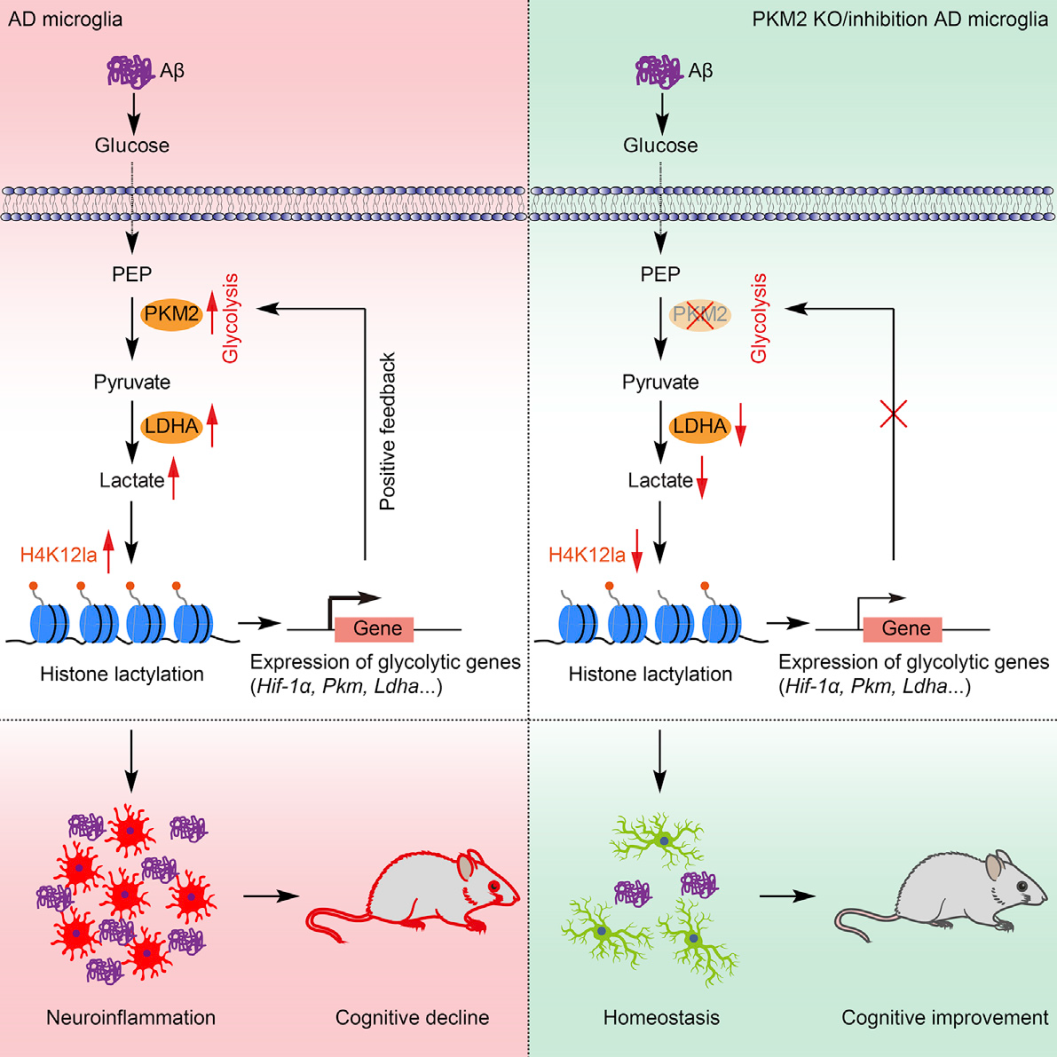
阿尔茨海默病组蛋白H4赖氨酸12乳酸化对小胶质细胞糖代谢的正反馈调节
实验方法:组蛋白提取,qChIP,海马细胞外通量测定,莫里斯水迷宫,经脑室(ICV)注射,硫黄素S (TS)染色,免疫荧光
小胶质细胞的促炎激活是阿尔茨海默病(AD)的一个标志,这一过程涉及从氧化磷酸化(OXPHOS)向糖酵解的转变。我们展示了小胶质细胞中的正反馈循环如何驱动AD的发病机制,并且我们证明抑制小胶质细胞中的这一循环可以改善AD小鼠模型(5XFAD)中的Aβ负担和认知缺陷。首先在5XFAD小鼠和AD患者的脑样本中检测到组蛋白乳酸化升高后,我们观察到H4K12la水平在Aβ斑块附近的小胶质细胞中升高。这种乳酸依赖性组蛋白修饰在糖酵解基因的启动子上富集并激活转录,从而增加糖酵解活性。最终,糖酵解/H4K12la/PKM2正反馈回路加重了AD的小胶质细胞功能障碍。药理抑制PKM2可减弱小胶质细胞的激活,而小胶质细胞特异性消融PKM2可改善AD小鼠的空间学习和记忆。因此,破坏正反馈回路可能是治疗AD的一种潜在的治疗方法。本文于2022年4月发表于Cell Metabolism (IF=31.373)。
技术路线:

结果:
(1) 遗传性AD模型小鼠海马乳酸水平升高
已知小胶质细胞具有代谢灵活性。鉴于AD小胶质细胞中从OXPHOS到糖酵解的代谢重编程,我们首先检测了5XFAD小鼠海马中的乳酸水平。5XFAD是一种成熟的AD转基因模型。比色分析显示,与年龄匹配的WT小鼠相比,12月龄5XFAD小鼠的乳酸水平显著升高(图1A),这与先前报道的结果一致,即AD患者的脑脊液(CSF)乳酸浓度高于健康对照组。

图1:5XFAD小鼠和AD患者组蛋白乳酸活性升高
(2) 5XFAD小鼠和AD患者组蛋白乳酸化升高
由于乳酸作为一种前体可以刺激组蛋白乳酸化,因此很容易假设组蛋白乳酸化可能在AD的背景下发生改变。酸提取组蛋白的WB分析显示,与年龄匹配的WT小鼠相比,12月龄5XFAD小鼠的额叶皮层和海马中的泛赖氨酸乳酸化(Pan Kla)和H4K12la水平均有所增加(图1B)。通过对小鼠的观察,AD患者死后脑组织中Pan Kla和H4K12la的水平较健康人明显升高(图1C)。这些结果支持在AD的情况下乳酸和组蛋白乳酸化都增加。
(3) H4K12la水平在5XFAD小鼠斑块邻近小胶质细胞中特异性升高
为了检测不同脑细胞类型中组蛋白乳酸化的变化,我们将H4K12la与针对神经元(NeuN)、星形胶质细胞(GFAP)和小胶质细胞(Iba1)标记物的抗体进行了免疫荧光共染色。大多数细胞显示H4K12la荧光(图1D-1F);在5XFAD和WT小鼠之间,我们没有观察到神经元或星形胶质细胞中H4K12la强度的明显差异(图1D和图1E)。对离体神经元和离体星形胶质细胞中酸提取的组蛋白进行WB检测显示,5XFAD小鼠和WT小鼠的H4K12la水平没有差异(图1D和1E)。然而,与远离斑块的5XFAD小胶质细胞和WT小胶质细胞相比,Aβ斑块邻近小胶质细胞的H4K12la信号强度显著升高(图1F)。我们从12个月大的5XFAD和WT小鼠的成年大脑中分离小胶质细胞,然后用组蛋白酸提取和抗多种形式组蛋白乳酸化的抗体进行WB分析,结果表明H4K12la是5XFAD小鼠斑块邻近小胶质细胞中最普遍的差异影响组蛋白乳酸化修饰(图1G)。
(4) 小胶质细胞中H4K12la转录结果的全基因组分析
组蛋白修饰改变可以影响靶基因的转录激活和抑制。为了探索H4K12la在AD小胶质细胞中的潜在功能意义,我们进行了全基因组CUT&Tag分析,以确定小胶质细胞中受H4K12la调控的候选基因。通过磁分离收集12月龄WT或5XFAD小鼠的总脑细胞或小胶质细胞(图2A):使用抗H4K12la抗体的CUT&Tag和deepTools分析显示,与WT相比,5XFAD大脑的小胶质细胞中H4K12la峰明显富集(图2B)。小胶质细胞对比显示14437个不同的H4K12la结合峰,其中41.33%的峰位于启动子序列(%3Kb)内。在所有细胞样本中,5XFAD小鼠中只有670个差异H4K12la结合峰,其中26.12%位于启动子序列(图2C)。
为了研究H4K12la对5XFAD小鼠小胶质细胞的表观遗传调控作用,我们将5XFAD小鼠小胶质细胞启动子处不同H4K12la结合峰的4410个靶基因按基因本体划分为不同的KEGG通路(图2D)。这些KEGG通路包括参与糖酵解和炎症的HIF-1、toll样受体、NF-kB和tnf信号通路(图2D)。这些峰在糖酵解基因(包括Hif-1a、Pkm和Ldha)上鉴定出候选基因组位点,表明这些基因启动子处的H4K12la水平升高(图2E)。
定量染色质免疫沉淀(qChIP)分析表明,与WT小胶质细胞相比,5XFAD小鼠分离的小胶质细胞中Hif-1a、Pkm和Ldha启动子上的H4K12la水平显著升高(图2F)。与此一致,qPCR显示,这些基因在5XFAD小鼠的小胶质细胞中的表达水平显著升高(图2G)。鉴于我们发现AD小鼠的小胶质细胞中H3K18la水平升高,我们使用qChIP(使用针对H3K18la的抗体)检测了WT和5XFAD小鼠分离的小胶质细胞。总之,H4K12la修饰激活了AD小胶质细胞中编码已知糖酵解酶的多个基因的转录。
我们在2、6和12个月大的5XFAD和WT小鼠分离的小胶质细胞的时间过程数据集中检测了组蛋白乳酸化水平。H4K12la水平在5XFAD小胶质细胞中以年龄依赖的方式增加。与年龄匹配的WT小胶质细胞相比,6月龄和12月龄的5XFAD小胶质细胞中Pan Kla和H4K12la的水平均明显升高(图S2A)。随着5XFAD小鼠小胶质细胞中AD发病机制的进展,Hif-1a、Pkm2和Ldha的表达也随之显著增加(图S2B)。考虑到组蛋白乳酸化依赖于糖酵解产生的底物的存在,并且考虑到我们发现H4K12la修饰促进糖酵解基因的表达,似乎有可能在H4K12la和糖酵解之间存在正反馈回路,传播我们在AD大脑小胶质细胞中观察到的代谢开关/重新连接(图2H)。
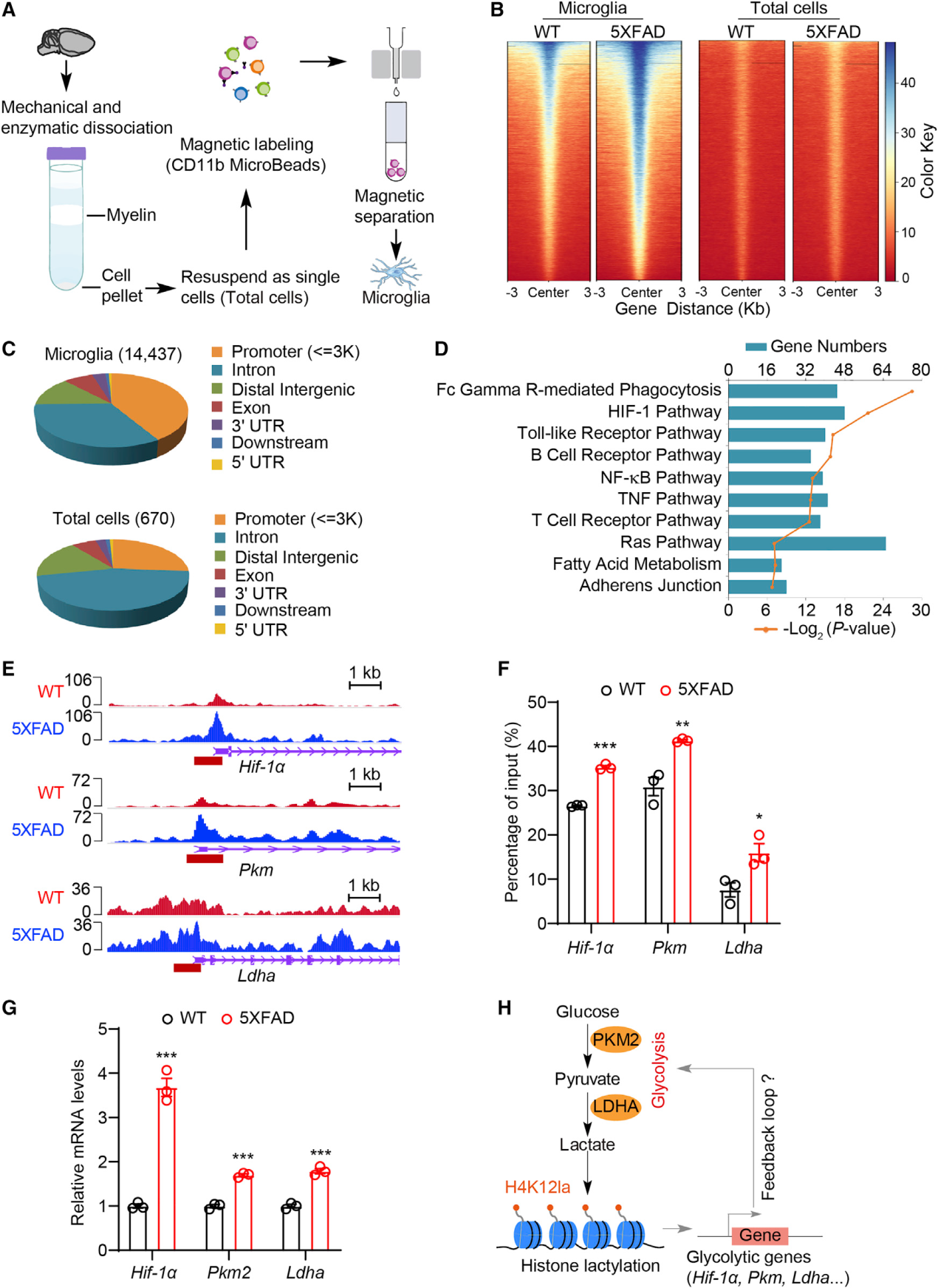
图2:小胶质细胞中H4K12la转录结果的全基因组分析
(5) 糖酵解/H4K12la/PKM2在AD小鼠小胶质细胞中形成正反馈回路
我们使用WB检测分离成人小胶质细胞中的PKM2水平,发现与年龄匹配的WT小鼠相比,6个月大的5XFAD小鼠中PKM2的小胶质细胞积累显著增加(图3A)。与PKM2的代谢功能一致,我们还发现AD小胶质细胞中的乳酸水平显著升高(图3B),支持AD小胶质细胞中有氧糖酵解的异常激活。
我们对小胶质细胞(抗iba1)和Aβ斑块区域(抗6e10)进行了PKM2免疫荧光染色。与我们的WB结果一致,该染色显示,与WT小鼠相比,AD小鼠脑切片中PKM2信号明显增加(图3C)。值得注意的是,PKM2的上调特异性地定位于Aβ斑块邻近的小胶质细胞,这与我们之前在AD小胶质细胞中检测到的H4K12la水平升高一致。PKM2与神经元标记物(抗neun)或星形胶质细胞标记物(抗gfap)共染色表明PKM2未与神经元共定位(图3D),但在5XFAD和WT脑的星形胶质细胞中均显示明显的PKM2信号(图3E);星形胶质细胞中糖酵解的基础水平较高,这一发现与之前的报道一致,即星形胶质细胞可以向附近的神经元供应乳酸。这些结果支持了AD小胶质细胞中存在一个由活性糖酵解、H4K12la和PKM2组成的正反馈循环的概念。
我们对死后人AD脑切片的PKM2和小胶质细胞标记物(抗iba1)和Aβ斑块(硫黄素S, TS)进行了免疫荧光染色。与我们在5XFAD小鼠中观察到的结果一致,与斑块远端和健康个体的小胶质细胞相比,PKM2在斑块邻近小胶质细胞中特异性上调(图3F)。总之,这些都支持糖酵解/H4K12la/PKM2正反馈回路可以“锁定”AD小胶质细胞的代谢重连接。
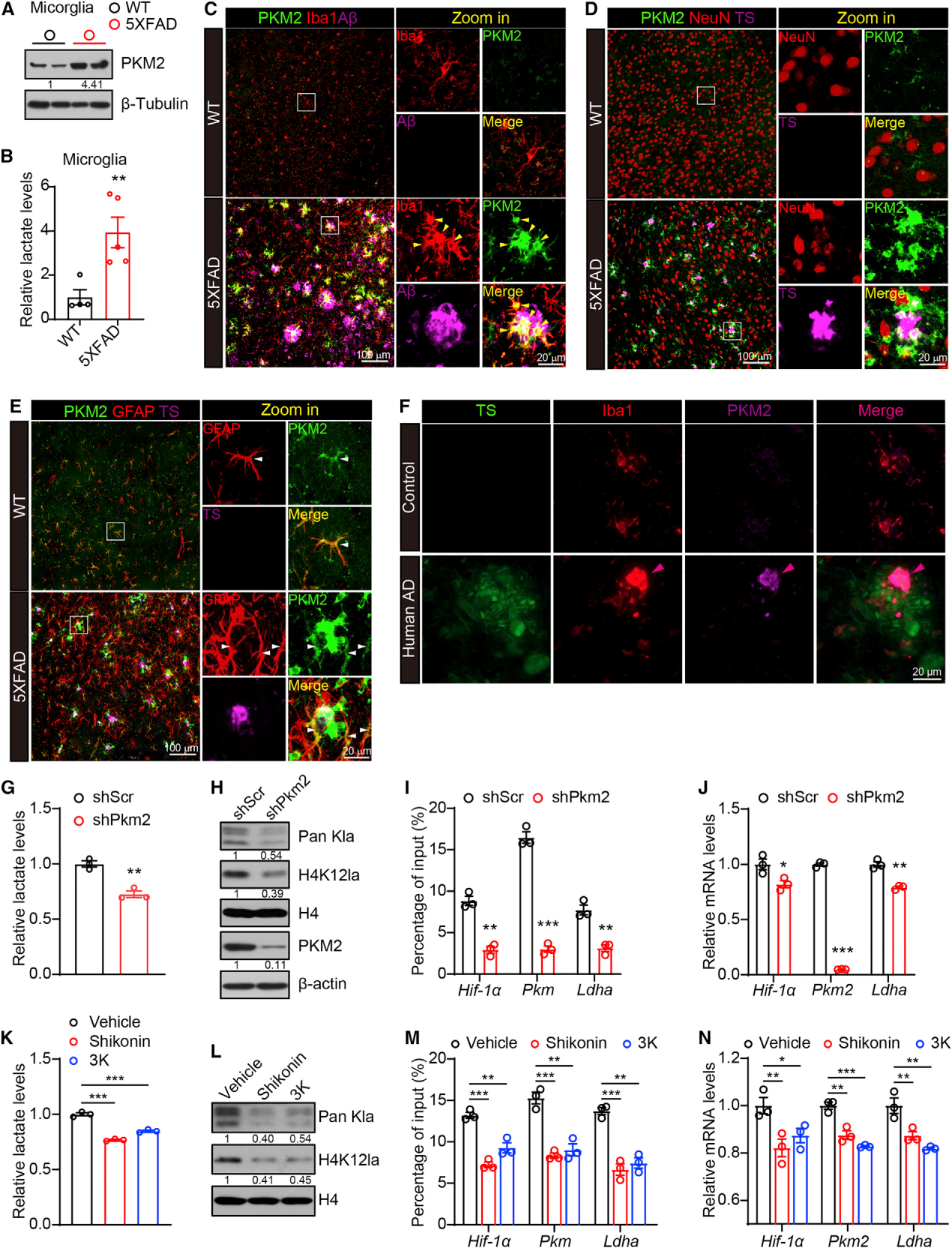
图3:糖酵解/H4K12la/PKM2在小胶质细胞中形成正反馈回路
(6) 抑制PKM2破坏小胶质细胞中的糖酵解/H4K12la/PKM2正反馈回路
为了实验证实糖酵解/H4K12la/PKM2环对小胶质细胞的功能影响,我们在培养的小胶质BV2细胞中构建了PKM2稳定敲除细胞系。Pkm2的敲低导致细胞内乳酸水平以及Pan Kla和H4K12la水平的显著降低(图3G和3H)。重要的是,qChIP分析显示,与对照细胞相比,Pkm2敲低的BV2细胞中糖酵解基因启动子(包括hif-1a、Pkm和Ldha)上的H4K12la水平显著降低(图3I)。与之前h4k12la介导的转录激活一致,qPCR显示,在Pkm2敲低的BV2细胞中,hif-1a、Pkm2和Ldha的表达水平显著降低(图3J)。
用已知的PKM2抑制剂(紫草素或化合物3K)治疗BV2细胞导致细胞内乳酸水平以及Pan Kla和H4K12la水平显著降低(图3K和3L)。与Pkm2敲低细胞的结果一致,紫草素或复方3K处理显著降低了hif-1a、Pkm和Ldha启动子上的H4K12la水平(图3M),这些基因的表达也显著降低(图3N)。总之, PKM2抑制破坏了小胶质细胞中的糖酵解/H4K12la/PKM2正反馈回路。
(7) 阻断PKM2阻断糖酵解/H4K12la/PKM2环可抑制5XFAD小鼠的小胶质细胞活化
我们通过将Pkm2f/f小鼠与CX3CR1CreER小鼠杂交(在5XFAD背景下),实现了小鼠小胶质细胞中Pkm2的特异性缺失(图4A)。为了消融小胶质细胞中的Pkm2,2月龄小鼠连续5天灌胃他莫昔芬20 mg。随后对5个月大的动物进行行为测试和病理分析(图4A)。PKM2水平在cKO、AD (Pkm2f/f;CX3CR1CreER;5XFAD)小胶质细胞与f/f;AD (Pkm2f/f;5XFAD)小胶质细胞的比较(图4B)。小胶质细胞Pkm2敲除导致PKM1水平和ATP产生显著增加(图S3)。与此一致的是,海马细胞外通量测定显示,与WT小胶质细胞相比,Pkm2敲除的小胶质细胞耗氧量(OCR)显著升高(图S3D)。这些结果表明,小胶质细胞Pkm2敲除导致PKM1的代偿性表达和糖酵解向OXPHOS的代谢转变。我们还检测到Pkm2敲除后的小胶质细胞细胞内乳酸水平、Pan Kla和H4K12la水平以及Hif-1a和Ldha的表达均显著降低,无论是体外还是体内(图4C和S4A-S4E)。注意,在添加乳酸盐后,这些下降又恢复了(图S4A-S4E)。额外的免疫荧光分析证实,Pkm2缺乏降低了小胶质细胞中H4K12la的水平,斑块邻近小胶质细胞的降低尤为明显(图4D)。
AD是一种慢性神经炎性疾病,伴有小胶质细胞异常激活,逐渐导致小胶质细胞功能障碍。我们用抗PKM2和抗Aβ(抗6E10)抗体对小胶质细胞(抗iba1)进行了共染色。AD脑切片中具有强PKM2信号的小胶质细胞异常激活;这些表型在cKO;AD脑切片中不明显(图4E)。WB显示,与f/f;AD提取物相比,cKO;AD海马提取物中Iba1水平明显降低,进一步支持小胶质细胞增生(图4F)。
对小鼠脑切片进行Iba1、Aβ(抗6E10)和TMEM119(一种小胶质细胞特异性稳态蛋白)的联合染色。正如预期的那样,Iba1+TMEM119−表明,f/f;AD小鼠的Aβ斑块邻近小胶质细胞偏离了稳态阶段小胶质细胞(图S5)。然而,Aβ斑块周围的小胶质细胞在cKO;AD小鼠中部分重新表达了TMEM119(图4G)。qPCR表明,与f/f;AD小胶质细胞相比,从cKO;AD小鼠中分离出的小胶质细胞显著增加了小胶质细胞稳态基因如Tmem119, P2ry12和Tgfb1的表达,而促炎基因包括Tnf-a, iNos和Il-1b的表达显著降低(图4H)。这些发现表明,通过基因删除PKM2来中断糖酵解/H4K12la/PKM2环可抑制5XFAD小鼠的小胶质细胞促炎激活。
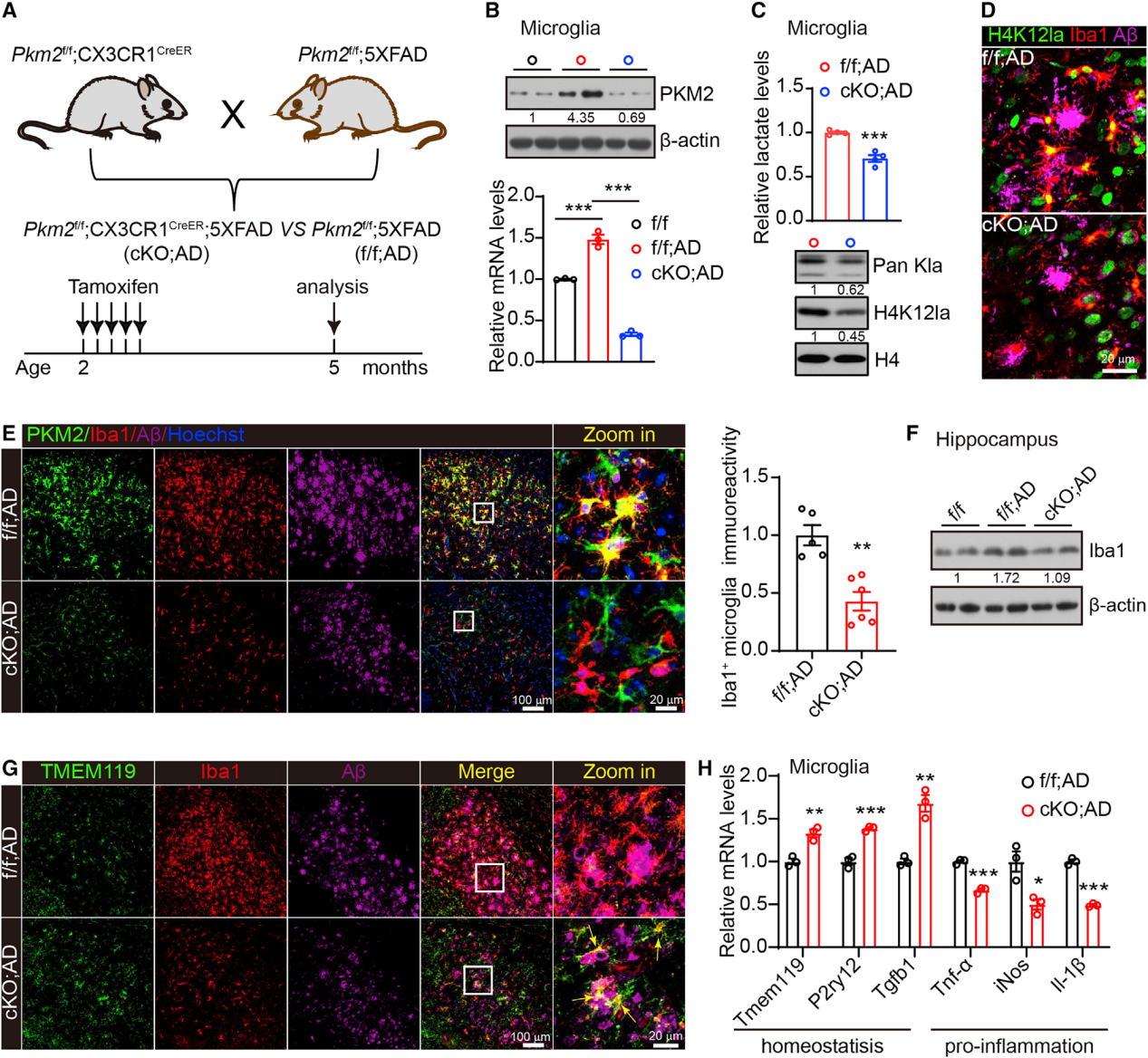
图4:PKM2基因缺失可改善5XFAD小鼠的小胶质细胞功能障碍和神经炎症
(8) PKM2特异性消融术可改善5XFAD小鼠的Aβ病理
小胶质细胞介导的神经炎症促进了Aβ的产生并促进了Aβ斑块的形成。我们采用TS染色法检测f/f;AD和cKO;AD小鼠脑切片中的Aβ斑块,探讨糖酵解/H4K12la/PKM2环的中断是否会影响AD小鼠的Aβ病理。小胶质细胞特异性消融Pkm2导致包括皮质和海马在内的大脑区域Aβ斑块含量显著降低(图5A和5B)。与我们的TS染色结果一致,WB显示cKO;AD小鼠的皮质和海马中Aβ水平与f/f;AD小鼠相比显著降低(图5C)。此外,与f/f;AD小鼠相比,cKO;AD小鼠营养不良神经突的总面积(lamp1阳性免疫染色信号)显著减少(图S6),表明AD小鼠小胶质细胞Pkm2缺失具有神经保护作用。这些结果表明,通过破坏PKM2功能来中断糖酵解/H4K12la/PKM2环可以改善5XFAD小鼠的Aβ病理。
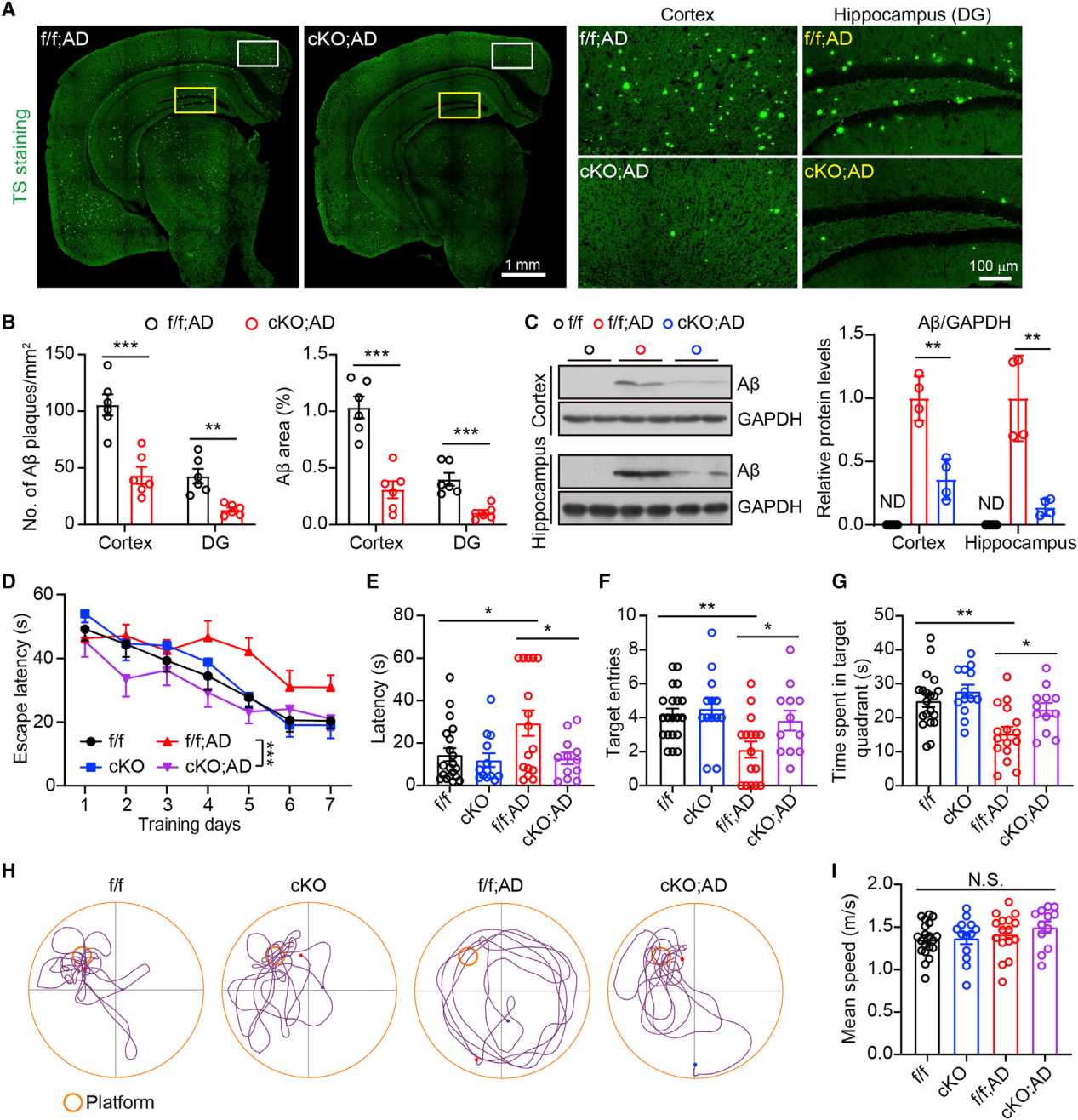
图5:5XFAD小鼠小胶质细胞中PKM2特异性消融可改善Aβ病理并改善空间学习和记忆
(9) PKM2特异性消融术可改善5XFAD小鼠的空间学习和记忆
Morris水迷宫实验检测空间学习记忆能力发现,与f/f;AD小鼠相比,cKO;AD小鼠的学习能力显著提高(即在训练试验中,cKO;AD小鼠对平台的逃避潜伏期更短;图5D)。cKO;AD小鼠在探针试验期间首次进入目标(平台)的潜伏期明显缩短,进入目标的次数明显增加,在目标象限的时间明显增加(图5E-5H),结果表明cKO;AD小鼠的记忆保持能力更好。值得注意的是,游泳速度在小鼠之间没有明显差异(图5I),这表明cKO;AD小鼠这些表型的减轻不是由于运动能力的改变。总的来说,通过阻断PKM2来中断糖酵解/H4K12la/PKM2环可以挽救5XFAD小鼠的空间学习和记忆缺陷。
(10) 药理抑制PKM2可降低5XFAD小鼠的Aβ负荷和小胶质细胞活化程度
为了测试药理学上抑制PKM2是否对AD有任何保护作用,我们每隔一天给4个月大的5XFAD小鼠脑室内注射PKM2抑制剂(紫草素或化合物3K),持续1个月,之后对大脑进行AD相关病理分析。PKM2的化学抑制显著降低了海马和皮质等脑区Aβ斑块的含量(图6A和6B)。此外,这两种抑制剂也明显降低PKM2水平,降低反应性小胶质细胞的程度,并部分逆转5XFAD小鼠小胶质细胞的变形虫形态(图6C和6D)。化学抑制PKM2也显著抑制了低聚抗体刺激的原代培养小胶质细胞中IL-6和TNF-a等促炎因子的水平(图6E)。总之,通过抑制PKM2功能来中断小胶质细胞中的糖酵解/H4K12la/PKM2环可以被理解为减轻AD神经炎症的潜在策略。

图6:药理抑制PKM2可减轻5XFAD小鼠的Aβ负荷和神经炎症程度
结论:糖酵解/H4K12la/PKM2正反馈回路加剧了AD的小胶质细胞激活和功能障碍。通过阻断PKM2阻断该环可降低H4K12la水平,从而在转录上抑制一组糖酵解基因,从而降低乳酸水平。除了我们证明这种抑制可以改善小胶质细胞功能障碍之外,我们的研究还表明,抑制这种恶性反馈循环代表了治疗AD的潜在治疗策略。
参考文献:Pan, R. Y., He, L., Zhang, J., Liu, X., Liao, Y., Gao, J., Liao, Y., Yan, Y., Li, Q., Zhou, X., Cheng, J., Xing, Q., Guan, F., Zhang, J., Sun, L., & Yuan, Z. (2022). Positive feedback regulation of microglial glucose metAβolism by histone H4 lysine 12 lactylation in Alzheimer's disease. Cell metAβolism, 34(4), 634–648.e6. https://doi.org/10.1016/j.cmet.2022.02.013