






剩饭新炒的Stomatin依旧可以发高分
实验方法:细胞培养及诱导成脂分化,BSA偶联棕榈酸的制备与处理,慢病毒包装和感染,脂质滴分离,脂质积累定量,WB,免疫荧光(IF),免疫共沉淀(IP),脂质组学分析,荧光漂白恢复(FRAP)实验,细胞表面生物素化,邻位连接技术(PLA),脂肪酸摄取实验,转基因小鼠构建,小鼠表型分析,葡萄糖耐量试验,病理学分析,转录组测序
脂肪酸摄取、脂质产生和储存以及脂滴代谢的调节与脂质稳态、脂肪细胞肥大和肥胖密切相关。本文发现stomatin是脂筏的主要成分,通过调节相关信号通路参与脂肪生成和脂肪细胞成熟;在脂肪细胞样细胞中,增加的stomatin通过促进LD-LD融合来促进LD的生长或扩大;stomatin还通过将效应分子(如FAT/CD36易位酶)募集到脂筏中来促进脂肪酸的内化,从而促进脂肪酸从细胞外环境中的摄取。高脂饮食喂食(HFD)的气孔蛋白转基因小鼠表现出肥胖、胰岛素抵抗和肝脏损伤;然而,在用常规饮食喂养的转基因动物中没有观察到这种表型;通过基因敲低或OB-1抑制stomatin通过下调PPARγ途径抑制脂肪分化和LD生长;stomatin对PPARγ的影响涉及ERK信号传导;然而,也可能存在其余途径。本研究于2022年7月发表在《Nature Communications》IF:17.694期刊。
技术路线:

主要实验结果:
1、脂肪形成过程中stomatin表达增加
为探究脂肪细胞分化,建立细胞模型:MD处理小鼠3T3-L1成纤维细胞3天,随后胰岛素处理第3 ~ 7天,使这些细胞分化为脂肪细胞样细胞,油红O染色可观察到细胞内脂质沉积(图1a)。这些细胞表达stomatin和主要脂肪细胞基因,如PPARγ,C/EBPα,Perilipin(图1b)。在第7天,这些脂肪样细胞中在DIC显微镜下可观察到大的脂质滴(LD);免疫荧光染色显示stomatin的亚细胞分布,除了细胞质中的点状染色外,在质膜(箭头所示)以及LD的表面也可见stomatin蛋白(图1c)。在STED显微镜下检查时,stomatin部分地与周围磷脂蛋白共定位(图1d)。当从脂肪样细胞中分离出LD时,LD中存在stomatin以及已知的LD相关蛋白磷脂蛋白(图1e)。
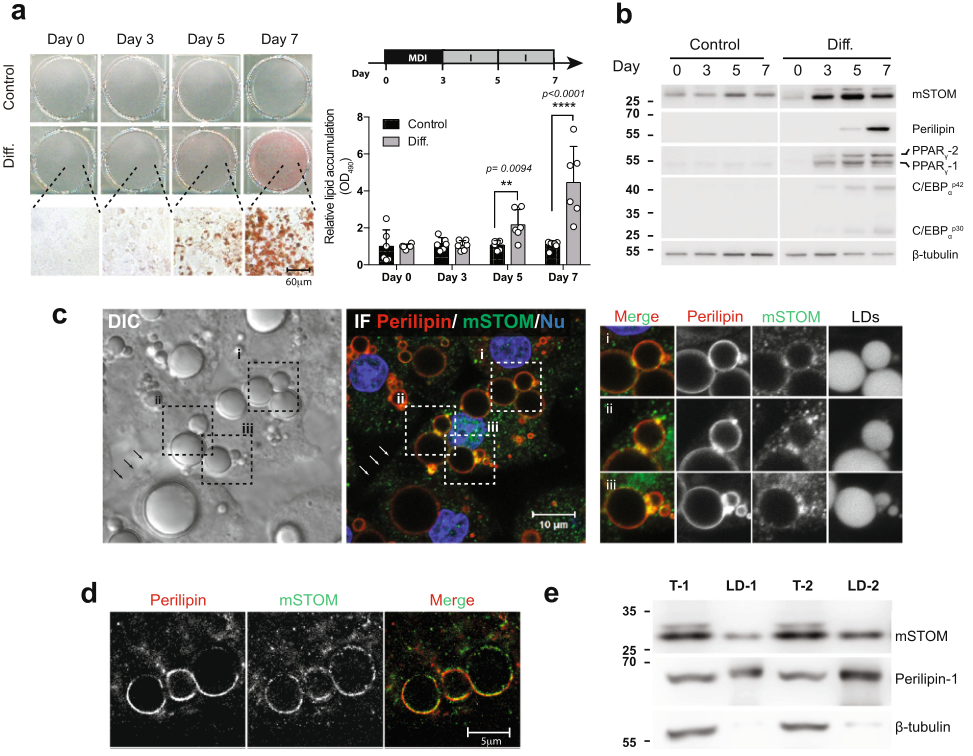
图1 在脂肪形成过程中,stomatin表达增加
2、高水平的stomatin促进LD增大
在小鼠3T3-L1细胞中过表达人stomatin基因(hSTOM),这些细胞被诱导分化为脂肪样细胞,结果显示过表达stomatin不影响脂肪分化,通过比较脂质积累(图2a)和脂肪基因的表达(图2b)。通过测定单个LD的大小,发现stomatin过表达细胞中LD更大(图2c)。通过两个小LD的融合可以形成一个大LD。延时记录表明在过度表达stomatin的脂肪细胞样细胞中存在这样的LD-LD融合事件(图2d)。
或者,大的LD可能是由较小的LD囊泡逐步“填充”脂质含量导致的,有些在光学显微镜下看不清。作者利用FRAP实验来研究这种可能性。如图2e所示,如图2e所示,在过表达stomatin的脂肪细胞样细胞中,LD的光漂白荧光含量导致了更快的荧光恢复(绿色,顶部痕迹),相比之下,对照细胞的荧光恢复要少得多、慢得多(蓝色,底部痕迹)。
在体外测量LD-LD融合事件(图2f,g)。从细胞中分离出LD,其LD被加载绿色Bodipy-FL-C16或红色Bodipy-558/568-C12荧光标记的脂肪酸类似物(箭头,图2g)。一些LD在体外混合时,似乎发生了融合,形成了黄色的LD(箭头,图2g),因为它们同时含有荧光的脂肪酸类似物。LD-LD融合的程度可以通过计算融合的LD百分比来量化。由于stomatin可以分泌到培养基中,作者注意到,与从对照组细胞收获的条件培养基(CM-Ctl)相比,添加从过表达stomatin的细胞中获得的条件培养基(CM-hSTOM)会导致融合的LDs百分比增加。有趣的是,通过用抗stomatin抗体处理条件培养基来消耗stomatin,有效地抑制了这种融合促进活性(图2g)。
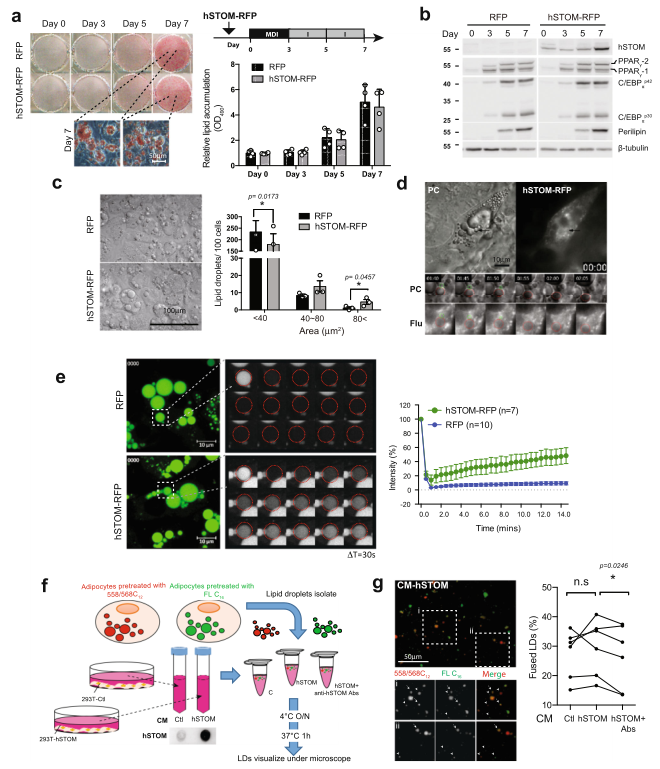
图2 stomatin过表达促进脂肪样细胞LD的生长和融合
3、高水平stomatin促进脂肪酸摄取并且stomatin被招募到质膜上与CD36相互作用
作者使用IP使用鉴定和stomatin结合的蛋白,共鉴定到885个候选蛋白,其中183个蛋白存在于质膜(PM),包括CD36和CAV1,它们与脂质储存相关。免疫沉淀实验证实stomatin与CD36的分子相互作用(图3a)。荧光标记的棕榈酸类似物Bodipy-FL-C16,当添加到培养基中时,被3T3-L1脂肪细胞样细胞内化。吸收过程可以通过测量细胞内荧光的增加来持续监测(图3b)。过表达stomatin的脂肪细胞样细胞(绿色)比对照组细胞(蓝色)表现出更多和更快的脂肪酸吸收。这样的效果不是由扩散引起的。在过表达stomatin(灰色痕迹)和对照组脂肪细胞样细胞(黑色痕迹)之间的Bodipy-FL摄取效率没有发现明显变化。
Stomatin不仅促进脂肪酸的吸收,而且在这个内化过程中从LD表面重新分布到质膜上。如图3c所示,当用BSA作为对照处理细胞时,在被检查的大约一半脂肪细胞样细胞中,stomatin蛋白以高丰度存在于LD周围。然而,当用BSA结合的棕榈酸(PA)处理这些细胞15分钟时,大多数与LD相关的stomatin似乎与LD "解离",出现在质膜上或分散在细胞膜上。
位于细胞表面的膜蛋白的数量可以用表面生物素化的方法进行标记,并与细胞内的蛋白分别进行量化。如图3d所示,计算了相对于总蛋白(输入),stomatin、CAV-1、CD36和ATP1A1的表面生物素化部分(表面)。只存在于细胞内部的肌动蛋白被用作对照。结果发现,与BSA处理相比,用PA处理细胞时,CAV-1、CD36和ATP1A1的表面部分仍然没有变化。相反,用PA处理脂肪细胞样细胞时,stomatin的表面部分比用BSA处理时有所增加(箭头)。这样的增加可能是由于PA的吸收引发了stomatin从LD或其他细胞内室被招募到质膜上,促进了来自细胞外环境的脂质成分的内化。
使用PLA进一步原位分析了stomatin和其他LD相关蛋白,如CD36或CAV-1之间的分子相互作用。如图3e所示,当用PA处理3T3-L1脂肪细胞样细胞时,PLA+位点显著增加,表明stomatin和CD36之间沿质膜分子结合(箭头)。撤消PA处理可以逆转这种stomatin-CD36的相互作用。相反,PA处理并不影响stomatin和CAV-1之间的相互作用(图3f)。
以上表明STOM招募到质膜上并与CD36相互作用
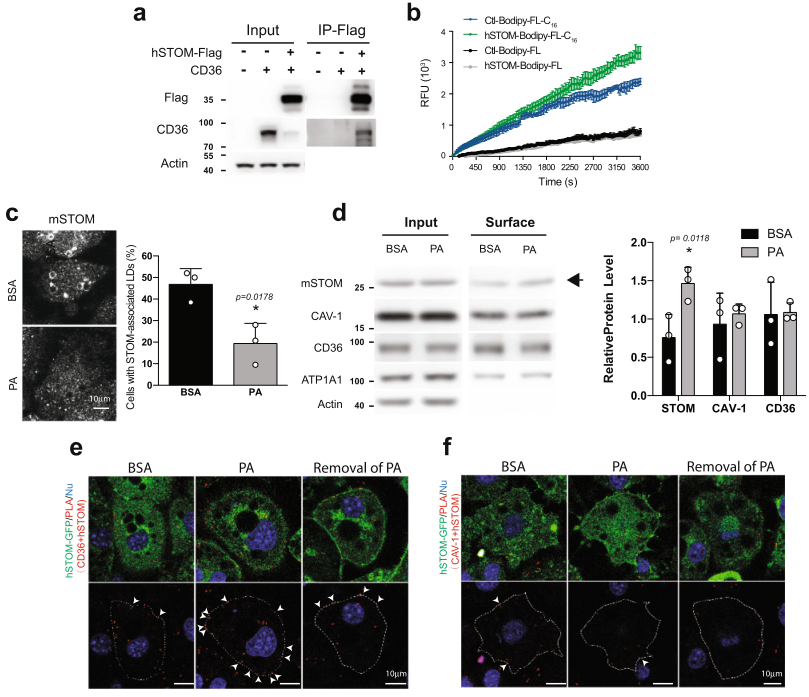
图3 stomatin与CD36互作促进脂肪酸摄取
4、STOM升高导致喂HFD小鼠肥胖
研究饮食对stomatin表达的影响。野生型小鼠(WT)用普通饲料(CD)或高脂肪饮食(HFD)喂养20周。收集脂肪垫,并通过qPCR检测stomatin(图4a)。与CD喂养的小鼠相比,HFD喂养的小鼠皮下(SAT)和内脏(VAT)白色脂肪组织中mSTOM的mRNA增加,但这种增加在棕色脂肪组织(BAT)中不太明显。
STOM转基因(STOM Tg)小鼠是通过将人类stomatin基因工程到动物体内产生的。这些小鼠在SAT中含有大量的hSTOM蛋白(图4b),这些蛋白主要在白色脂肪细胞的表面膜上(图4c)。STOM Tg和对照组WT小鼠自3周岁起用CD或HFD喂养20周;每周对它们进行称重(图4d)。当用CD喂养时,STOM Tg与WT小鼠的体重增加相似。然而,当用HFD喂养时,STOM Tg小鼠比WT更快地增加体重(图4d,e)。在HFD喂养20周后,STOM Tg小鼠的体重比WT小鼠至少高17%。全身成分测量显示,脂肪的增加比瘦肉、自由液体或总水的增加更明显(平均增加32%)(图4f)。
体内脂肪酸摄取实验是通过将BSA乳化的Bodipy-FL-C16注射到小鼠的尾静脉,孵化15分钟,然后测量动物白色脂肪组织中的荧光信号(图4g)。观察到到STOM Tg小鼠比WT小鼠有更多的脂肪酸吸收到脂肪组织中(图4g)。
以上表明,在体内stomatin转基因小鼠脂肪摄取更多更快,小鼠更肥胖。
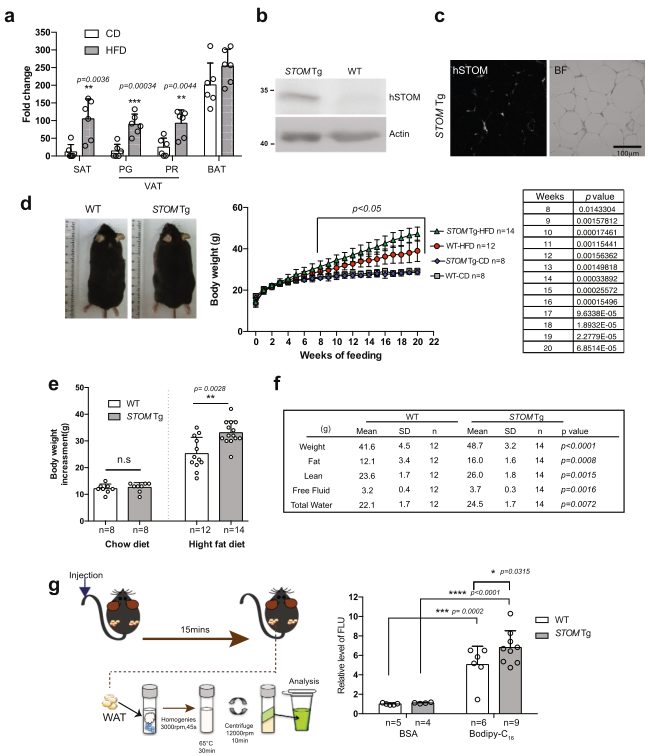
图4相比于对照小鼠高脂饮食喂养的stomatin转基因小鼠更加肥胖
5、HFD喂养stomatin转基因小鼠脂肪细胞肥大与代谢紊乱
经过20周的HFD喂养,具有上调stomatin的小鼠显示出比WT小鼠更多的SAT和BAT的质量,而他们的VAT组织相对保持不变(图5a)。组织学分析显示,在HFD喂养的STOM Tg小鼠中,SAT的脂肪细胞出现肥大,并且比WT小鼠的脂肪细胞大小更大(图5b)。如图5c所示,虽然STOM Tg和WT小鼠的空腹血糖水平相似,但通过HOMA-IR实验测得的血浆胰岛素和胰岛素抵抗明显升高,通过腹腔葡萄糖耐量试验(IPTGG)测得的葡萄糖耐量在HFD喂养的STOM Tg比对照组小鼠更难耐受。肥胖通常与肝脏中的异位脂肪堆积有关。事实上,HFD喂养的STOM Tg小鼠表现出更大的肝脏质量(肝脏肿大),以及大泡和小泡的脂肪变性。这些表型与肝功能受损有关,血清中GPT和GOP水平的升高就是证明(图5d)。

图5 高脂饮食喂养的stomatin转基因小鼠表现出脂肪细胞肥大与代谢紊乱
6、敲低STOM阻断脂肪生成和LD变大
将两种针对小鼠stomatin基因不同位点的shRNA分别包装在慢病毒颗粒中并导入3T3-L1细胞,得到shSTOM-1和shSTOM-2细胞。两种shRNAs都能有效地下调stomatin的表达。敲除stomatin抑制了脂肪生成,这一点从诱导分化后缺乏脂质积累得到证明(图6a)。参与脂肪细胞分化的基因的表达,如PPARγ和C/EBPα,因stomatin敲除而减少(图6b)。STOM的下调也抑制了LD的成熟和生长(图6c)。
比较stomatin敲低和对照组3T3-L1脂肪细胞样细胞的转录组谱。共鉴定到379个转录物有显著差异,包括185个上调的基因和194个下调的基因。使用转录组分析控制台(TAC)将这些基因映射到128条Wikipathways上,排名靠前的富集途径如图6d示。在"脂肪生成基因 "通路中,涉及该通路的12个基因显著差异,其中9个(75%)被下调,3个(25%)被上调(图6e)。为进一步验证芯片结果,进行qPCR实验,重点是脂肪生成相关基因Pparg、Cebpa、Dlk-1、Fabp4和Cfd基因(图6f);除了Dlk-1,所有这些基因都被下调了。

图6降低stomatin表达影响脂肪生成并抑制脂滴生长
7、STOM通过ERK通路调节脂肪形成
如图7a所示,敲除stomatin导致PPARγ的减少。Akt途径没有受到STOM敲除的影响,而ERK途径被激活(箭头)。脂肪生成基因C/EBPβ和C/EBPδ是PPARγ调节的上游。在诱导脂肪生成分化后,C/EBPβ和C/EBPδ,在前三天表现出短暂的增加,随后从第三天到第七天逐渐减少(图7b-c)。在stomatin敲除的3T3-L1脂肪细胞样细胞中,发现stomatin的缺乏在第7天引起C/EBPβ的减少(图7b)。相比之下,stomatin敲除能够维持或增加C/EBPδ的表达,使之达到比对照组高得多的水平(图7c)。
ERK途径的激活已被证明可抑制PPARγ,从而禁止脂肪细胞的分化和脂肪生成。U0126是一种高度选择性的ERK抑制剂。用U0126处理shSTOM-1脂肪细胞样细胞,理论上U0126可能减轻stomatin敲低对pERK激活的影响。然而,U0126处理并没有扭转hSTOM-1细胞的脂质积累不足的表型(图7d)。相反,用PPARγ激动剂TGZ处理shSTOM-1细胞,能够部分挽救脂质积累缺陷;有趣的是,U0126和TGZ的双重处理可以进一步促进脂质积累(图7e)。这些结果表明,stomatin存在一种目前未知的机制,可以正向调节PPARγ并激活脂肪生成,而不依赖于ERK。

图7 敲低Stomatin激活ERK通路
Stomatin的功能抑制可以通过药理试剂OB-138实现,该试剂通过干扰Stomatin的自我结合,抑制Stomatin的活性。首先确定了OB-1的LD50(图8a)。用OB-1处理3T3-L1脂肪细胞样细胞,以剂量依赖的方式有效减少了脂质积累(图8b)。OB-1处理后,LD的大小也减少(图8c),磷酸化ERK的水平增加(图8d)。
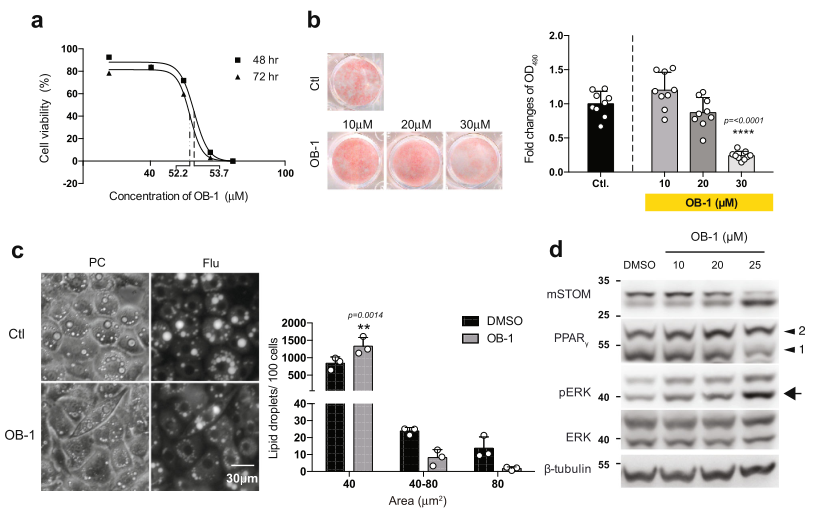
图8 Stomatin抑制剂OB-1抑制脂肪生成,阻碍脂滴生长
总之,提出了stomatin在调节成脂分化和调节LD生长和脂肪细胞摄取脂肪酸中的作用的工作模型(图9a)。
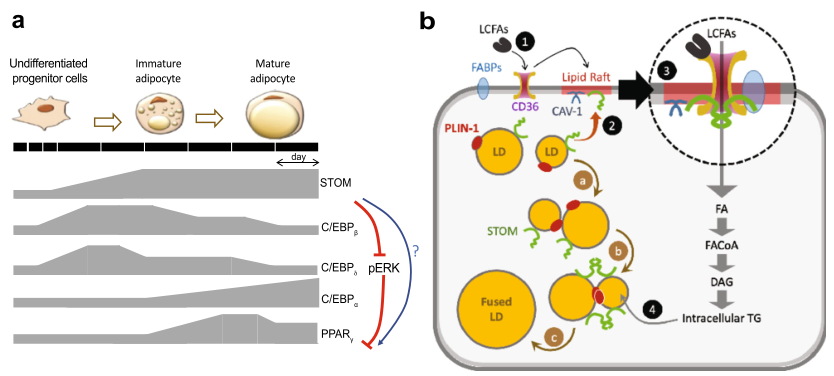
图9图解模型说明了诱导成脂分化过程中动态基因表达和stomatin在促进脂肪细胞脂肪酸摄取和脂滴增大中的作用
参考文献:
Wu SC, Lo YM, Lee JH, Chen CY, Chen TW, Liu HW, Lian WN, Hua K, Liao CC, Lin WJ, Yang CY, Tung CY, Lin CH. Stomatin modulates adipogenesis through the ERK pathway and regulates fatty acid uptake and lipid droplet growth. Nat Commun. 2022 Jul 19;13(1):4174. doi: 10.1038/s41467-022-31825-z. PMID: 35854007